| |
VIII./4. fejezet: Szekvenciadatok előállítása újgenerációs szekvenálással
Az újgenerációs szekvenálás munkafolyamata a szekvenálási (wet lab szakasz), és az adatelemzés (in silico elemzés, bioinformatika) szakaszokra osztható.

2. ábra Az újgenerációs szekvenálás munkafolyamatának áttekintése
|
|
 |
VIII./4.1. Könyvtárkészítés
A nukleinsav megfelelő eljárással történő kivonása a biológiai mintából kritikus lépés, amely a végső szekvenálási eredményekre is jelentős hatással van. Különösen az olyan speciális minták esetén kell körültekintően eljárni, amelyekben a DNS alacsony koncentrációban, vagy töredezett formában nyerhető ki (szövettani feldolgozások átesett (szakirodalomban formalin-fixált-paraffinba ágyazott: FFPE, formalin-fixed, paraffin-embedded)) minták gyakran izomszövet, daganatszövet).
Az izolálást követően szekvenálás első laboratóriumban történő lépése a könyvtárkészítés, mely magában foglalhatja a DNS-fragmentálást, a célszekvencia feldúsítását és amplifikációját, a fragmentumok méret szerinti kiválogatását és több minőségellenőrzési lépést, továbbá a DNS-fragmentumok „ragadós” végeinek kijavítását, molekuláris vonalkódolásukat (barcoding) és a szekvenáló adapter ligálását.
A fragmentálás során a kivont DNS-t enzimatikus vagy fizikai módszerekkel (centrifugálással, porlasztással vagy ultrahanggal) kisebb méretű fragmensekre darabolják. Ezt a lépést a kapott fragmentumok túlnyúló végeinek tompa végekre történő kijavításán, azon foszforilációján, az adott esetben különböző eredetű, az elemzés során megkülönböztetni kívánt fragmensek molekuláris vonalkódoláson (barcoding) adapter-oligonukleotidok ligálásán mennek keresztül, amit a tovább vinni szándékolt fragmensek méret alapján történő kiválasztása követ. A fragmentumok méret szerinti kiválogatására azért van szükség, mert az optimális és hatékony szekvenáláshoz a fragmentumok méretének meg kell felelnie az alkalmazott NGS-platform számára optimális hosszának.
A vizsgálni kívánt genomi szegmensek szelektív kinyerése (genom partícionálás, target enrichment) nagymértékben csökkenti a költségeket és a befektetett energia mennyiségét, miközben a növeli a szekvenálás hatékonyságát. A target enrichment technikák segítségével egyetlen gén, releváns gének csoportjai (panelek) vagy akár a teljes, fehérjét kódoló exoni szakaszok is feldúsíthatóak a mérésre szánt DNS-könyvtárban. A jelenleg a legszélesebb körben használatos technikák alapvető reakcióelvük alapján csoportosíthatók: a hybrid capture, amikor a mintából előállított DNS-könyvtárat az előre elkészített, a kiválasztott célrégióval komplementer, specifikus DNS fragmensekhez hibridizáltatják, ezáltal különíthetőek el a vizsgálandó szekvenciák a nem analizálandóktól. A hibridizáció után a partner nélkül maradt egyszálú fragmenseket lemossák, majd a vizsgálni kívánt DNS darabokra nézve feldúsított minta ezután eluálható. A nagyléptékű vizsgálatok során (több száz exont lefedő hasonló számú amplikontól a teljes exome-ig) a folyamat megvalósulhat szilárd lapka-hordozón (microarray) vagy oldatban (in solution, gyöngyök felszínére rögzített, a célszekvenciákkal komplementer oligonukleotidok segítségével) is. A módszerek eltérő technikai paraméterei miatt az alkalmazási területeik is nagymértékben különbözhetnek: ha összességében több megabázisnyi szegmens elemzése a cél, akkor a hibridizációs megközelítés a célravezetőbb, mivel nagyobb célrégiót képes átfogni, viszont gyengébb erősítést lehet a módszerrel elérni. Ezzel szemben, amikor kisebb a vizsgálandó célrégió, a PCR-alapú megközelítés előnyösebb (ld. a néhány gén lefedésére fejlesztett multigén-panelek), mivel nagy szekvenálási mélységet és gyakran egyenletesebb lefedettséget biztosít.
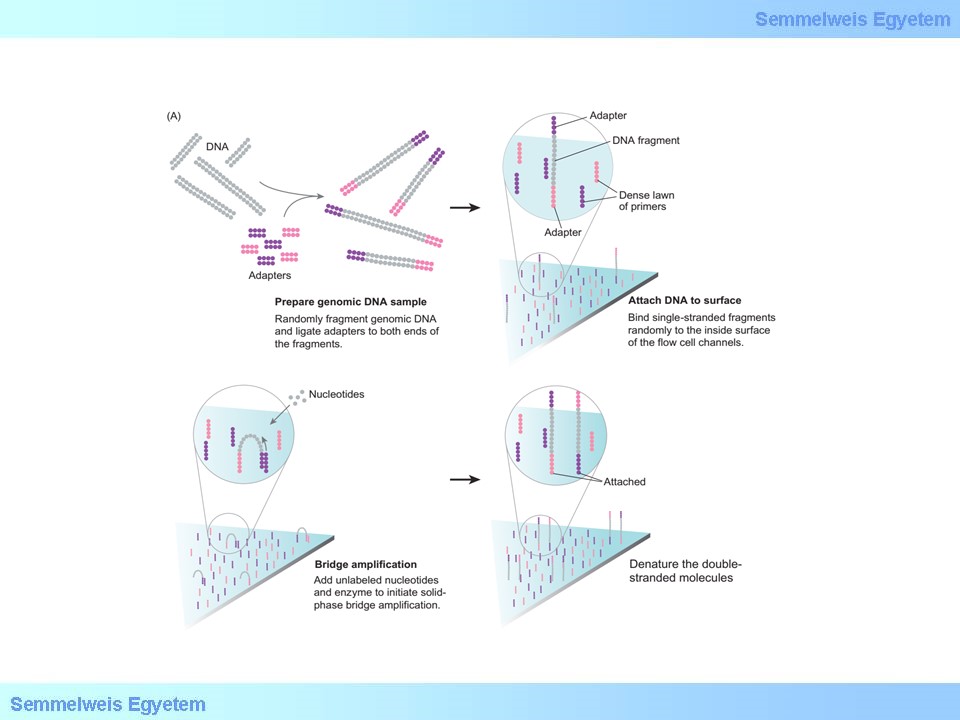
3.ábra: Könyvtárkészítés és klonális amplifikáció az Illumina szekvenálási platformokon
|
|
|
VIII./4.2. Klonális amplifikáció és a bázisok sorrendjének leolvasása
A különböző NGS-technológiák körében általános eljárás a templát szilárd felülethez vagy hordozóhoz kötése, így a DNS klonális amplifikációja is felülethez kötötten megy végbe. A templátról készült több ezer kópia egy meghatározott területen biztosítja a nukleotid beépülését kísérő szignál megkülönböztethetőségét a háttérzajtól a szekvenáló reakció során. Az NGS módszerek fő jellemvonását, a masszívan párhuzamosított leolvasások lehetőségét az egyetlen fragmens-templát pontos másolataival rendelkező egyedi reakciócentrumok biztosítják, amelyek egyidejű nyomonkövetése több millió DNS-kódrészlet párhuzamos szekvenálását teszi lehetővé.
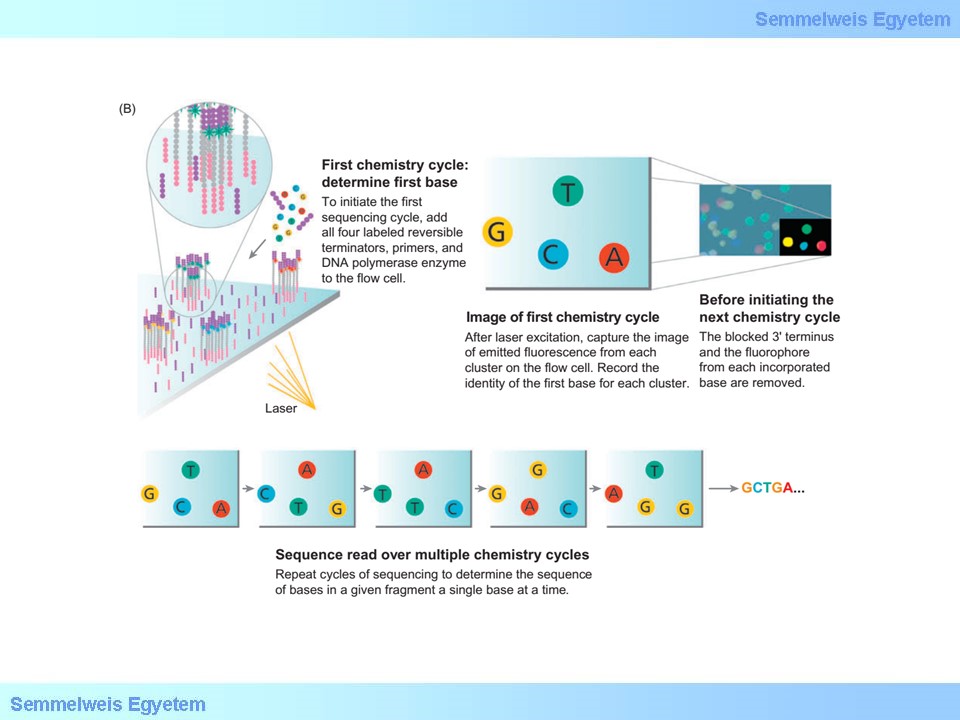
4.ábra A bázissorrend meghatározásának méréstechnológiája az Illumina szekvenálási platformokon (Annual Review of Genomics and Human Genetics by ANNUAL REVIEWS, 2008.)
|
Az NGS futtatás különböző platformokon valósulhat meg. A nagy teljesítményű NGS szekvenátorok esetében szinte mindig százas nagyságrendű az egy időben futtatott individuális minták száma. A klinikai gyakorlatban jelenleg jellemzően páros végű (paired-end) azaz mindkét irányból leolvasott, 150-300 bp méretű rövid leolvasott szakaszok (short reads) keletkeznek a mérés során. Számos platform került kifejlesztésre, a bázissorrend leolvasásában különböző kreatív megoldásokkal és persze eltérő hibakarakterisztikával. Az NGS méréstechnika műszaki megoldásai főként az egyedi bázisok leolvasásának módszerében térnek el (a piroszekvenálástól a jelenleg elterjedt fluoreszcens jelöléseken át a félvezető technológiával vagy nanopórus segítségével történő detektálásig).
|
|