| |
II./2.3.: Primary cerebral tumor
II./2.3.1.: Primary cerebral tumors, in general
|
 |
The preparation demonstrates the macroscopic features of one of the most important forms of the processes that reduce intracranial (ic) space: in this case it is a neoplastic process, a primary cerebral tumor, that acquires space for itself. Tumors occurring within the cerebral skull can be divided into two main groups (that is also true for the tumors of the spinal cord): the tumors evolving from the nervous tissue (primary), and the metastatic (secondary) tumors that arrive from other tissues. Those tumors that directly invade the cerebral tissue from its environment are discussed in other chapters of pathology.
The average occurrence of primary brain tumors is 7-10/100 000 habitant. They are especially frequent among children (from all the tumors, primary brain tumors are the second most frequent in this age group), and there is another peak in its frequency between the ages of 45 and 70 years. Their pathophysiology is unclear in most cases, however, in certain types the role of heredity (the so called tumor syndromes), environmental harms (development of meningeal tumors after the irradiation of the head-neck region), or the state of the immune system (association between the primary cerebral lymphomas [mainly B cell] and AIDS) are known.
II./2.3.2.: The classification of primary brain tumors
|
 |
Majority of the primary brain tumors origin from neural ectoderm. Primary central nervous system (CNS) lymphomas, the tumors of the dura mater and arachnoidea are involved also in this group, but these are not discussed here. In 1926, Percival Bailey and Harvey Cushing introduced the first classification of neuroectodermal tumors. These two outstanding researchers applied cyto- and histogenetical approach in the classification of tumors. They hypothesized that the elements of the tumors would suggest the source cell type to a certain extent, dependent on the level of their differentiation.
Thus, it seemed obvious that the most important tumors can be divided into two main groups:
-
- Glial tumors (gliomas): astrocytoma, oligodendroglioma, ependymoma;
and
-
- Tumors originating from a nerve cell („ganglion-cell” tumors).
In the development of brain tumors they suggested that the (cytogenetically) early developmental cell forms (spongioblast, medulloblast, astroblast, etc.) play a special role.
With the improvement of neurosurgery techniques, more and more types of tumors could be identified, and it has also become clear that in many cases tumors are not composed of only one cell type, but show elements similar to multiple cell types (mixed gliomas [e.g. oligoastrocytoma], glial-neuronal tumors [e.g. ganglioglioma]). The classification has evolved through a long and devious process and the details have rather historical significance by now.
|
| |
The current, officially accepted morphological classification is a rating system presented by the WHO in 2007, which differentiates more than hundred tumor types and subtypes. The evolution of the histological classification was simplified by the Kernohan-grading to a certain extent. Kernohan, who was a medical doctor at the Mayo Clinic (Rochester, MN, USA), took over his collegue’s, Albert Broders’s hypothesis that had been published in 1920, and according to which the clinical behavior (biological potential) of a tumor could be described most easily with the help of a numeric scale.
Broders divided the tumors originating from the mucous membrane of the mouth into 4 groups (grades).
-
- If tumor cells are similar to the original intact mucous membrane cells in >75% (highly differentiated form), then the tumor is in grade I (Gr I.).
-
- When similarity is 50-75% then the tumor is in grade II (Gr II.).
-
- Grade III is defined by 25-50% presence of differentiated cells (Gr III.)
-
- While the hardly differentiated, thus, significantly anaplastic tumors, in which <25% of the cells are similar to the cells constituting the original tissue/epithelium are in grade IV (GrIV).
Kernohan’s theory, according to which this system can mechanically be adapted to the tumors of the nervous system, proved wrong. The four grades based on the differentiation level can be applied – with modifications, and not automatically– mostly to astrocyte tumors, while among oligodendrogliomas and ependymomas only grades II and III can be found. The original theory has also been repeatedly modified. Currently, for example, cell types (phenotypes) that have no normal cell-type versions in the nervous system are part of the grading system (e.g. pilocyte, pylocitic astrocytoma). Contrary to the original Kernohan-theory, the current histopathological evaluation takes into account not only the similarity between the tumor cells and the original tissue cells but numerous other factors too, both cytomorphological and tissue related. From these, the differentiation between „focal” (GrI.) - astrocytoma pilocyticum – and „diffuse” tumors (i.e. without definitive border; practically all the rest of gliomas) – first described in the WHO grading in 2000 - has a special biological-clinical significance. It is important that in the separation of the different tumor types, the tumor’s biological behavior is now part of the classification. Astrocytic gliomas are the easiest to demonstrate this on. (Table 1.).
|
Look into the table!
|
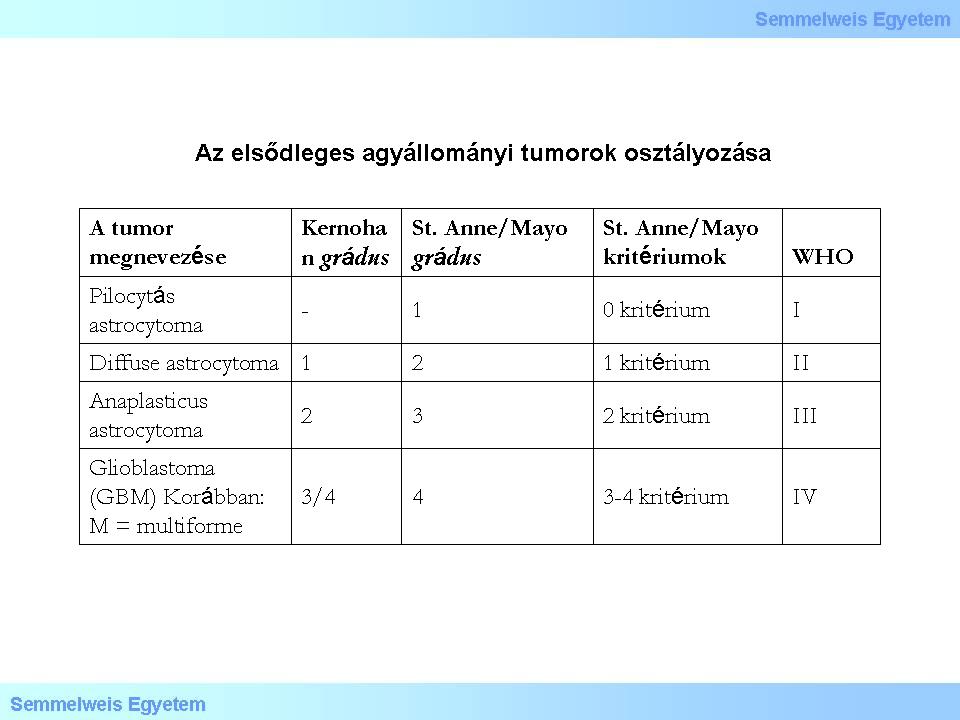
Table 1.
|
The prognosis of primary brain tumors is highly dependent on cytologic and tissue characteristics, as well as on molecular abnormalities accompanying or preceding the tumors. Among these, the most well-known factor is the significance of the „maturation” of the tumor cells (i.e. the cytologic differentiation). In the past 5-10 years, however, molecular/genetic lesions and cytogenetic deviations started to get more into the focus of attention. The evaluation of the level of differentiation, the so called grade of the tumor( from the latin word gradus), is hampered by the extreme histological heterogeneity within most of the individual tumors. For example, a glioma, highly differentiated at the borders might frequently hide poorly differentiated, anaplastic tumor, a glioblastoma (GBM) in its deep, hardly accessible parts. Despite all the difficulties, the classification of tumors is based on their histological characteristics because of their tight prognostic connections. In case of the most studied astrocytomas for example the determination of the level of differentiation is based on four criteria:
-
1) nuclear atypia,
-
2) cytologic heterogeneity (cytologic pleomorphism),
-
3) proliferation activity,
-
4) glomeruloid hyperplastic/necrotic endothelial cells surrounded by tumor cells (this fence-leth like configuration of tumor cells [they surround necrotic tissue in most of the cases] is called pseudopalisading).
With the help of these criteria four differentiation grades are determined:
-
- GrI (grade I) – if the tumor does not show any criterion;
-
- GrII – if the tumor shows at least one criterion;
-
- GrIII – if the tumor shows two criteria;
-
- GrIV – if the tumor shows three criteria.
This grading system had provided originally the base for the so called Saint Anne-Mayo classification. Although it later has changed several times along the WHO classification systems, the underlying principles can be accepted even today. The biological behavior of the glial tumors are mainly determined by their intensive infiltrational tendency. In these days, grade I tumors are hardly ever diagnosed and they are characterized by an „expansive” growth style. In contrast, diffuse growth style is typical for all the tumors from higher grades.
The anatomical position of the tumor is another important factor that substantially
determines the clinical course. It can show diagnostic value in case of tumors which are typcial for a certain anatomical site (site predilection). However, localisation has been proved to determine the tumor’s molecular „fingerprint” too: a frontal lobe GBM can show different characteristics from phenotypically the same GBM in the occipital lobe! Furthermore, due to other circumstances, histologically benign, highly differentiated tumors can cause death in the short term (for example meningeomas in the posterior cranial fossa, highly differentiated ependymomas in the base of the fourth ventricule, etc.). All tumors cause reduction of the intracranial space which in turn results in increased intracranial pressure.
Over time, and the course of tumor growth, the tumor tissue almost always dedifferentiates. However, it is typical that only in exceptional cases do intracranial tumors, even dedifferentiated ones, give extracranial metastasis.; their biological agressivity lies in their pronunced recurring tendency. The classification of the primary CNS tumors is currently based on the system published by the WHO in 2007, although this system is already out-of-date by now.
II./2.3.3.: Oncogenesis of the primary brain tumors
|
 |
The naming of the tumors suggest that they originate from some definitely committed cell form. Typical example for this is the name „oligodendroglioma”. It brings up several theoretical questions, first of all, whether this tumor really originates from an oligodendrocyte. It has, however, turned out that in certain areas of the fully developed brain there are pluripotent (progenitor) cells capable of proliferation. A part of these cells definitely play a role in the glial and neuroectodermal oncogenesis. Today we already know that DNA damages open the door for cellular mechanisms that could have never been imagined before: mesenchymal-epithelial/epithelial-mesenchymal transformation (MET and EMT), or the so called regressive differentiation. The latter means retrograde transformation of committed cells, in light of which, the bizarre possibility of the dedifferentiation of an oligodendrocyte due to genetic damage and thus, leading to the development of a tumor, cannot be ruled out. The current viewpoint of the possible pathways of glioma oncogenesis can be demonstrated on a figure taken from a recent (2010) publication (Figure 1.) Dashed arrows refer to the fact that there are still lots of uncertainties in this field. Additionally, it is noteworthy that the possible pathways of ependymoma oncogenesis cannot be found on the figure!
|
Look into the flowchart!
|
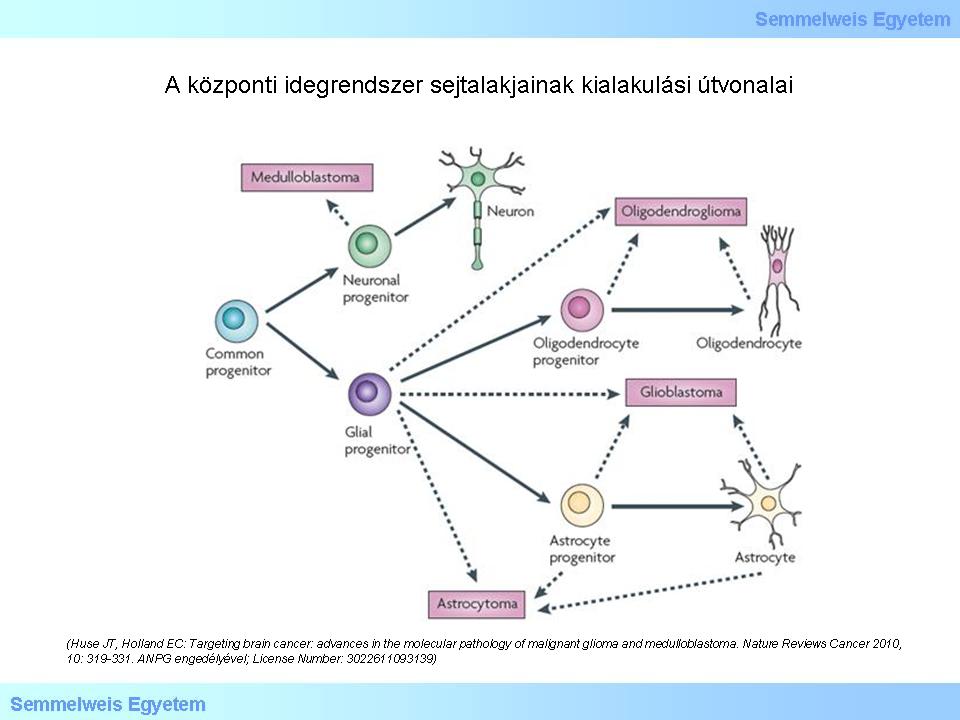
Figure 1.: Pathways of development of different cell types in the central nervous system (CNS). (Huse JT, Holland EC: Targeting brain cancer: advances in the molecular pathology of malignant glioma and medulloblastoma. Nature Reviews Cancer 2010, 10: 319-331. With the permission of ANPG; License Number: 3022611093139)
|
According to the theory presented on the figure, the cells differentiating to different directions originate from a common progenitor cell. In case of a genetic damage during the process of differentiation starting from the pluripotent cell (not a „stem cell” in the word’s real meaning), unlimited proliferation („immortalisation”) can be the result that leads to tumor development. Modern genomical analysis has shed light on those molecular pathways that can be responsible for the genesis of most of the gliomas. The report on the study of the GBM genom was published in 2008. It became clear that gene mutation of the receptor kinases (RTKs), such as the epithelial growth factor (EGFR) and the platelet-derived growth factor (PDGFR) play an important role in the glioma oncogenesis. The contribution of the growth factors to oncogenesis takes effect through the PI3K-AKT-mTOR and the RAS-MAPK signal transduction systems. It can be observed in the figure below that the damages in the p53 and RB tumor-suppressor pathways are also significant. The gene damage leads to abnormal proteins. Identifying the abnormal proteins provides the possibility to separate the different subtypes of gliomas (mostly of glioblastoma). In Figure 2 EGFR is indicated with red, PDGFR is indicated with blue and NF1 is indicated with green color. It can also be observed that other molecules known from oncology and normal proliferation cycle play an important role in tumor genesis too.
|
Analyze what you see on the figure!
|
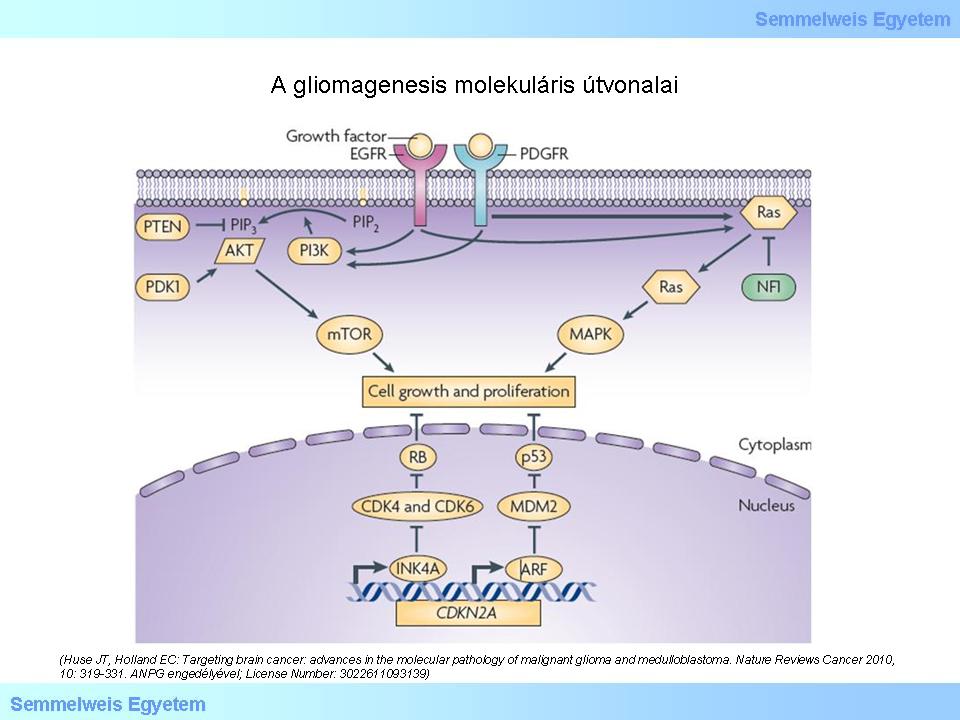
Figure 2: Molecular pathways of glioma genesis. CDK: cyclin-dependent kinase. NF1: neurofibromin. PDK1: 3-phosphoinositide-dependent protein kinase. PIP2: phosphatidilinositol-4,5-biphosphate. PIP3: phosphatidilinositol-3,4,5-triphosphate. (Huse JT, Holland EC: Targeting brain cancer: advances in the molecular pathology of malignant glioma and medulloblastoma. Nature Reviews Cancer 2010, 10: 319-331. With the permission of ANPG; License Number: 3022611093139)
|
The „Blue book” of WHO 2007 has already predicted the significance of these pathways. This schematic figure below (Figure 3) shows one of the pathways as classic „genetic/biological progression” (GrII. – GrIII. – GrIV.), typical for secondary glioblastoma. Primary GBM tumor groups follow other pathways.
|
|
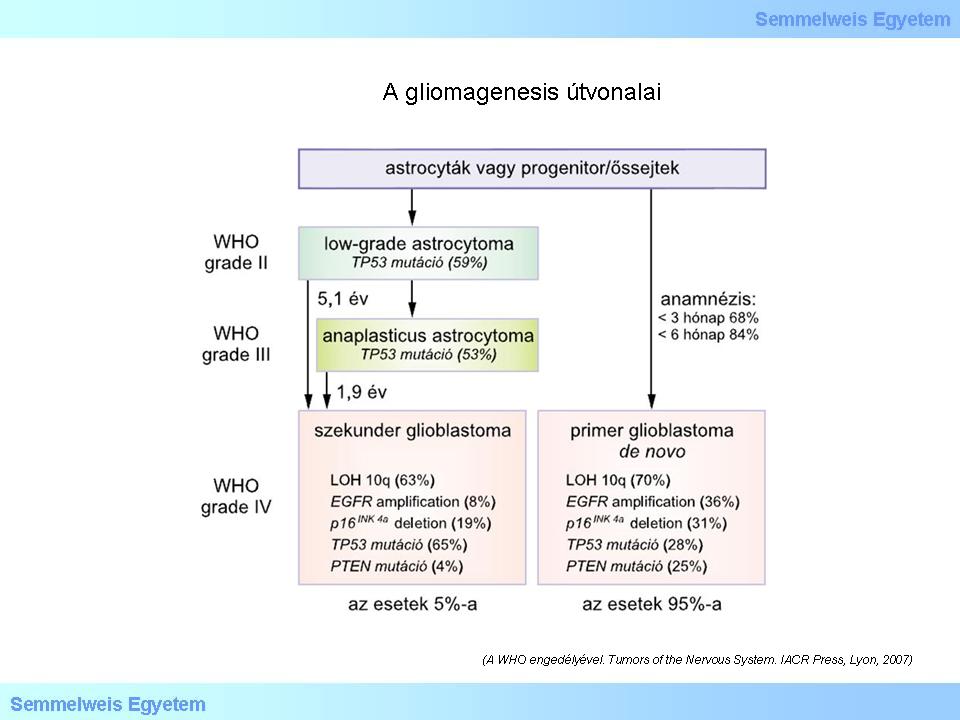
Figure 3.: Pathways of glioma genesis. (With the permission of WHO Tumors of the Nervous System. IACR Press, Lyon, 2007).
|
During the process of detailed mapping of genetic pathways, it turned out –practically as a „byproduct” - that in most gliomas the primary and common abnormity presents in the gene of a metabolic enzyme (isocitrate dehydrogenase-1: IDH-1). IDH-1is a cytoplasmic enzyme, and the missense point mutation happens at R132 localisation (this is the active center of the enzyme).
Today it is already known that although rarely, but the point mutation might also occur in the R172 location of the mitochondrial IDH-2 enzyme gene. The IDH-1/IDH2 mutations are typical in the early developmental phase of lower grade gliomas. If this mutation is followed by the deletion/co-deletion of 1p19q gene part, then the tumor acts, and in most but not all cases, looks like an oligodendroglioma. If the second step is a P53 mutation, then anaplastic (GrIII.) astrocytoma, later secondary glioblastoma (GrIV. GBM) develops. The significance of the IDH mutation lies in this enzyme’s fundamental role in lipid synthesis, in defense mechanisms against oxidative stress, in oxidative cell breathing, and in oxigene-sensory signal transduction.
|
|
Currently there is still a debate on through what mechanism the metabolite (2HG = 2-hidroxiglutarate) of the IDH-1 catalyzed process has an oncogenic effect and might result in brain tumors and acute leukemia in adults. Today, the immune histochemical examination of the presence of the mutant IDH-1 protein is already mandatory during the diagnostic process. In the case if the appearance of the tumor suggests the presence of oligodendroglial cells, FISH (fluorescence in situ hybridization) examination of the 1p19q is also mandatory. Based on the genetic studies, grade IV gliomas (glioblastomas) are now separated into four subtypes. These subtypes have prognostic value for patient survival. The degree of methylation of the promoter region of the 1p19q LOH and the MGMT (metilguanin-metiltranspherase) enzyme genes are predictive markers for whether what medical treatment the tumor is likely to respond to (in case of the 1p19q LOH it is the PCV, while in case of MGMT methylation alkilizer [temozolomide,Temodal] is suggested). However, in depth discussion of the relevant details is beyond the limits of this chapter (Table 2).
|
Look into the table!
|

Table 2: According to the current theory, there are four subtypes of malignant glioma: (1) proneural; (2) mesenchymal; (3) classic; (4) neural. The molecular pathogenesis and the clnical course of these subtypes are different. Studies targeting the details of this field are in the frontline of current neuro-oncopathology. (Huse JT, Holland EC: Targeting brain cancer: advances in the molecular pathology of malignant glioma and medulloblastoma. Nature Reviews Cancer 2010, 10: 319-331. With the permission of ANPG; License Number: 3022611093139)
|
II./2.3.4.: Astrocytomas
II./2.3.4.1.: General characteristics
|
 |
Astrocytomas can occur in any localisation and age. Majority of them has an infiltrative growing tendency, and they cannot be removed totally (in toto). The marginal, reactive astrocytosis can hardly be differentiated from the neoplastic astrocyte proliferation, even with the most developed diagnostic techniques. Thus, the border of the neoplastic and reactive parts is blurred, in many cases undifferentiatedly. In these cases sometimes the IDH-1 immune histochemical examination might help (see above). The intraoperative application of fluorescent ink might also help to successfully separate the neoplastic and intact parts. The treatment of astrocytomas are hampered by the modification of the blood-brain (blood-tumor) barrier in the way that the water-soluble chemotherapeutic agents can not cross and get into the tumor tissue. This is especially true for the well-differentiated tumor area types. Unfortunately, glial tumors are frequently not sensitive to radiation either. All in all: surgical intervention cannot remove the tumor totally, and neither chemotherapeutic agents, nor radiation are effective in the final eradication, leading to the poor prognosis of this type of tumor. The mean survival time of the most severe (and unfortunately very frequently occurring) type of diffuse malignant gliomas – the glioblastoma – is less, than 12-14 months even in case of the most up-to date treatment methods. In this regard, the rapidly developing identification of the molecular „fingerprints” of gliomas has brought a breakthrough. In those cases where both the IDH-1/IDH2 mutation is present and the promoter region of the NGNT enzyme gene is methylated, the combination of chemo- and radiotherapy (the so called Stupp protocol) results in even 2-4 years of patient survival, in growing number of cases!
II./2.3.4.2.: Highly differentiated (low grade; GrI.) astrocytomas
|
|
In many cases they do not need any radical treatment. They grow slowly and are symptom free for a long time, depending on the localisation. Cerebellar astrocytomas and childhood optic nerve and optic chiasm originated astrocytic gliomas are parts of this tumor group. The sample for the primary histologic diagnosis is usually taken by ultrasound or CT-guided stereotactic biopsy. The GrI. astrocytomas are so called pilocytic tumors. In case of the cerebellar pilocytic astrocytoma, its saccular character can be observed pathognomically and with imaging techniques too, specifically, that the tumor mass grows into the cavity of the cyst („mural nod”). In its impression smear, relatively monomorphic, long, spindle-like, bipolar cells can be observed with long cell processes, round and oval nuclei containing fine-clotted chromatin. There is no considerable cell- or nuclei polymorphism. Part of these tumors can be totally removed surgically, in which cases the prognosis is excellent. (Note: Based on the guidelines published by the WHO in 2007, morphological abnormalities in pilocytic astrocytomas disturbingly overlap with the characteristics of GBM – the diagnosis is far from simple despite what might be expected from the descriptions simplified due to didactic reasons!)
II./2.3.4.3.: Diffuse (medium/well differentiated; GrII.) astrocytomas
In this group (15P-1. macro-picture; 15P-1. micro-picture) fibrillar, protoplasmic and gemistocytic histological forms are differentiated. An important characeristic of these tumors is their tendency to oncologically progrediate (graduate reduction of the level of differentiation, dedifferentiation). This type of clinical/biological/molecular progression must not be mistaken for the radiological progression! The first is the continuous accumulation of genetic damage, while the latter is the tumor’s growth in size.
|
Look into the pictures!
|
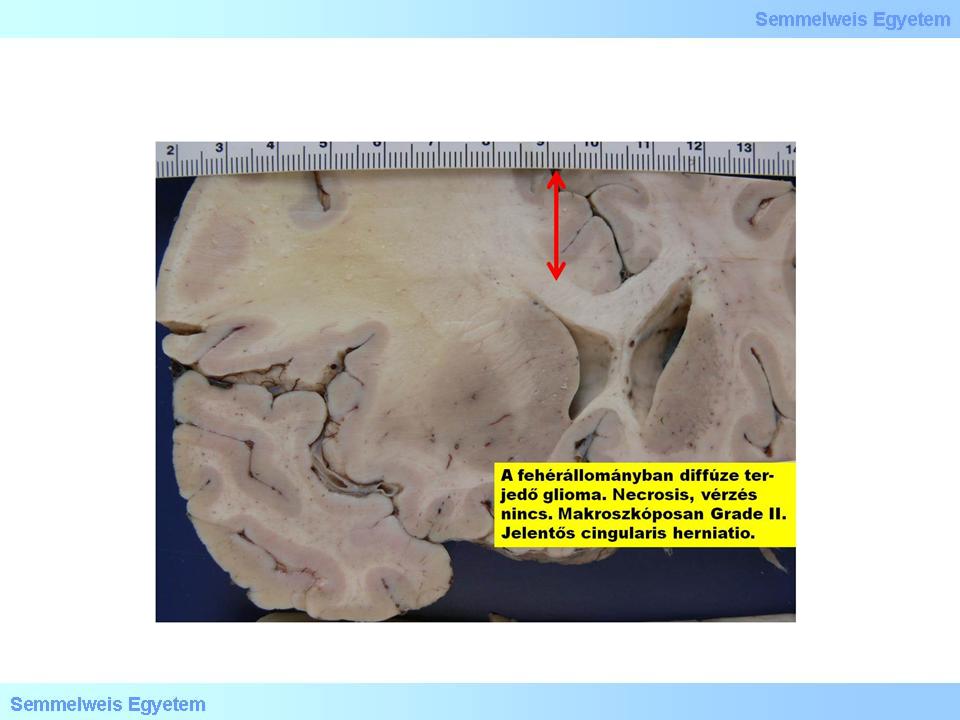
Macrograph 1: Diffuse glioma in the white matter. There is no necrosis, haemorrhage, that is why it is graded as Gr.II based on the macroscopic appearance, contrary to a Gr.IV glioblastoma which would be colourful because of these factors. Significant cingular herniation and midline (see the red arrow!) deviation. (A favor of Péter Molnár; University of Debrecen, Medical and Health Sciences Center, Department of Pathology)
|
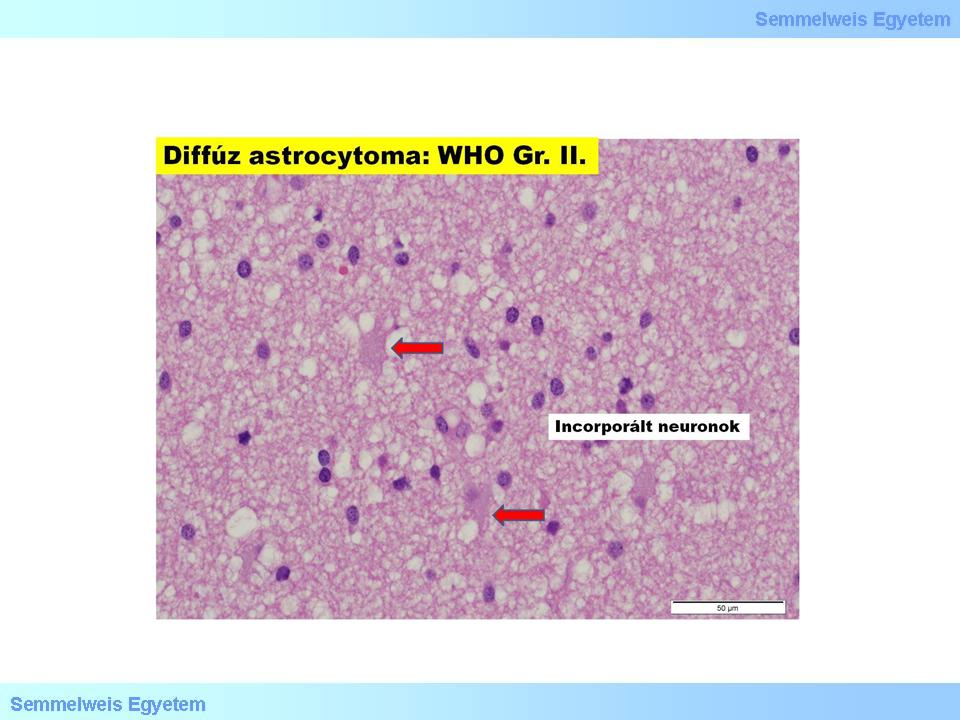
Micrograph 1: Diffuse astrocytoma (GrII.). (A favor of Péter Molnár; University of Debrecen, Medical and Health Sciences Center, Department of Pathology)
|
During the biological progression, the tumor continuously deviates from its original appearance histologically, its malignancy and biological agressivity increases. This is how a Gr.II tumor becomes first Gr.III (anaplastic astrocytoma), then Gr.IV (glioblastoma multiforme). In its microscopic appearance, fibrillary astrocytoma is characterized by minimally increased cellularity, moderate variability of the cells, moderate nuclear hyperchromasia, at the same time, structural disorganisation, and infiltrative growing of the tumor are typical. In case of possible, more pronounced nuclear atypia, grading does not increase. Mitotic cell is not, or just rarely observable. There is no necrosis and endothelial proliferation.
The diagnostic criterion for gemistocytic astrocytoma is that at least 20% of tumor cells have to be gemistocytes. These cells are in fact swollen astrocytes that can appear with a reactive nature in acute and chronic brain disturbances, and with a neoplastic nature in this subtype of astrocytomas. Gemistocytes have ample, eosinophilic cytoplasm, and eccentric nucleus (it is said that the name originates from the german word: „gemästet”=swallen), in between the cells the bacground matrix is coarse-fibrillary. Cellularity is moderately increased (15P-2A. micrograph). Cells show intensive positivity for anti-GFAP immune histochemical reaction (15P-2B. micrograph). With electron microscope, mass intermediate filaments and often megamitochondria are characteristic for these cells. They are typically tend to progrediate, and this can happen explosively, the time of which cannot be predicted based on the morphology.
|
Look into the pictures!
|
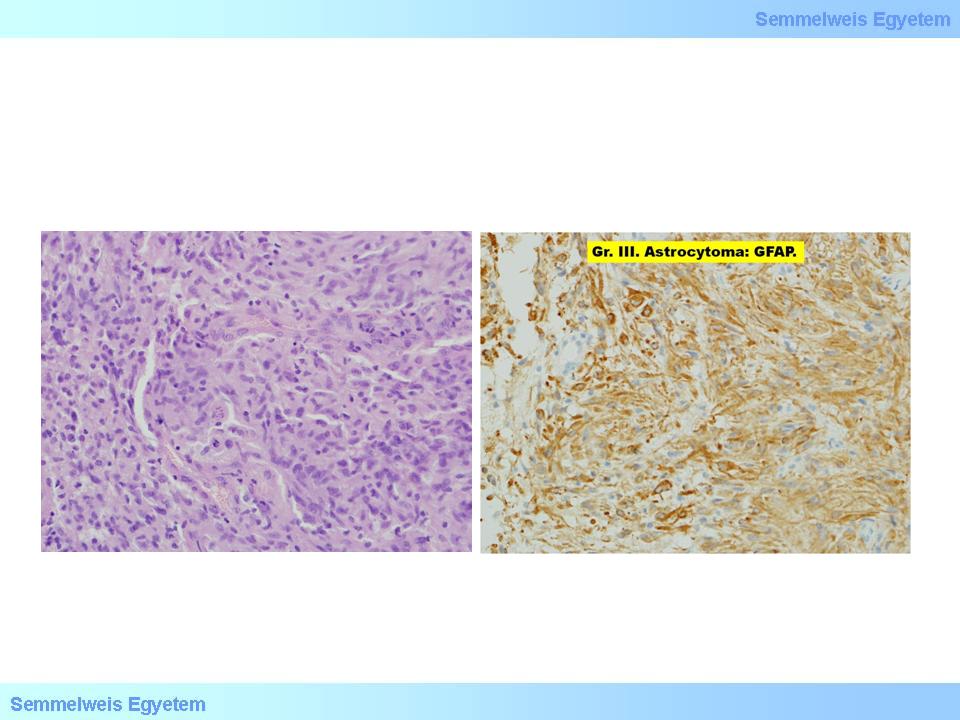
2A-B. micrographs: A: Astrocytoma – in the middle two big cells (gemistocyte). B: The tumor shows strong GFAP-positivity. (A favor of Péter Molnár; University of Debrecen, Medical and Health Sciences Center, Department of Pathology)
|
II./2.3.4.4.: Anaplastic astrocytomas (GrIII.)
Histologically, anaplastic astrocytomas are characterized by the intense cellularity of the diffusely growing tumor, the expressive nuclear atypia and hyperchromasia, and the increased mitotic activity. Heterogeneity, typical for astrocytomas in general, is strikingly present: shape- size- and painting differences of the cytoplasm and the nucleus, as well as the elevated mitotic rate are all expressed. For the up-to-date quantification of mitotic activity the automatic evaluation (x/1000 cells) of the phosphohiston 3 (PPH3) immune histochemical reaction is the most appropriate. Less reliable method is the application of antibody (Mib-1) produced against Ki-67. Over time, with the progression the histological picture becomes more and more agressive, cell forms with multiple nuclei, bizarre nuclei start to be present, while within the nuclei, inclusions and prominent nucleoli can be observed, sporadically also gemistocytes can be present.
In this group of tumors there is no necrosis, endothelial proliferation, glomerulal capillar proliferation (to sum up, there is no „microvascular proliferation”: MVP). Compact cellgroups, often different from their environment to a certain extent, might indicate the formation of clonogenic subpopulations. The reason for the initiation of clonal selection of the tumor cell population (that is the survival of the more viable cell groups in the given microenvironmental milieu) is yet unkown. During its process, the selected clones overgrow the tumor cell types around them, which are more differentiated but slower in reproduction, untill the least differentiated tumor (glioblastoma multiforme) stands alone, leading to the complete dedifferentiation of the tumor, that is, in fact, the oncological progression.
II./2.3.4.5.: Glioblastoma (multiforme) (GB[M], GrIV.)
This is taken as the most malignant tumor of the central nervous system. Without treatment, the survival time is only a few months (6-9) and even with the most up-to date, combined treatment it cannot be pushed over 10-14 months. To the naked eye, the sharp border of the tumor is misleading (macrograph 2), because it is, in fact, an infiltrative tumor, and despite the sharp contours, we do not deal with a metastasis. Because of its tendency for extensive necrosis that can lead to extensive parenchymal haemorrhage due to the abnormal vascularization of the tumor tissue, the macroscopic picture often mimics intracerebral haemorrhage (3A-C. macrograph), while the clinical picture shows the symptoms of stroke.
|
Analyse the features seen on the pictures!
|
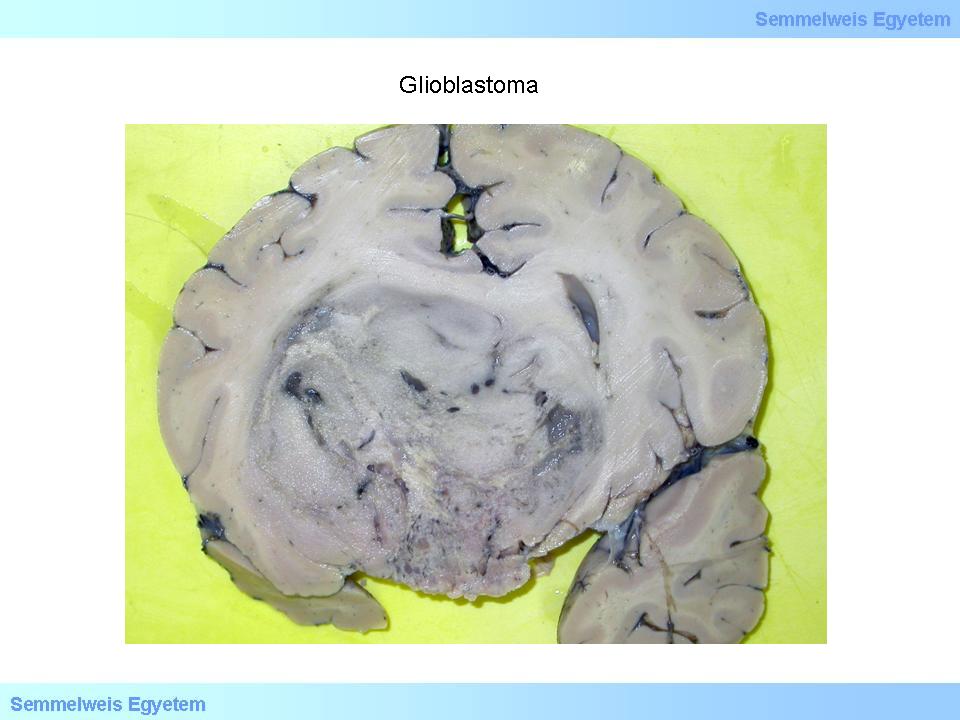
Macrograph 2: The macroscopically sharp borders of glioblastoma are confusing since this tumor is histologically infiltrative. This specific tumor mass caused serious cerebral deformation. (A favor of Péter Molnár; University of Debrecen, Medical and Health Sciences Center, Department of Pathology)
|
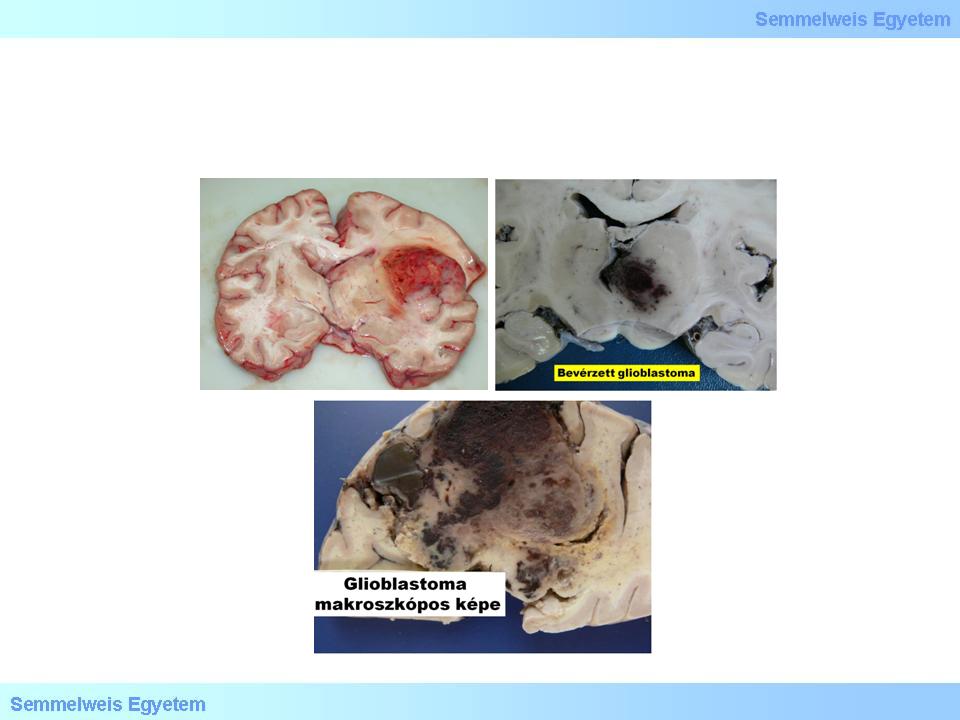
3A-C. Macrographs: For its haemorrhagic tendency, the macroscopic picture of the cut surface of a glioblastoma multiforme is colourful, often mimics stroke (intracerebral haemorrhage). (A favor of Péter Molnár; University of Debrecen, Medical and Health Sciences Center, Department of Pathology)
|
This tumor has primary and secondary forms. The primary glioblastoma multiforme develops de novo, while the secondary GBM is a result of the progression of lower grade astrocytomas. The glioblastoma (multiforme – we rarely use this adjective anymore since monorph glioblastoma [GB] subtypes are already known, such as the so called microcytaer glioblastoma) is a result of either the proliferation of pluripotent progenitor cells, or the progressive dedifferentiation of more differentiated tumor forms. Diagnostic criteria suggested by the WHO-2007 are histological: necrosis with surrounding pseudopalisading (fence-leth like configuration of tumor cells) and/or MVP. In addition, cytologic deviations described in anaplastic astrocytomas can also be found. The vital tumor tissue is hypercellular, cells are anaplastic, undifferentiated (micrograph 3), the background is spongiform (microcystic) or fibrillary.
|
|

Micrograph 3: Microscopic picture of glioblastoma szöveti képe: anisonucleosis, hyperchromasia, endothelproliferation. (A favor of Péter Molnár; University of Debrecen, Medical and Health Sciences Center, Department of Pathology)
|
II./2.3.5.: Oligodendrogliomas
II./2.3.5.1.: Grade II oligodendrogliomas (OGDG, GrII)
It is macroscopically an often calciferous tumor in typically hemispherial localisation, developing dominantly in the cortex and subcortical white matter, and infiltrating extensively the cortex. In the microscopic picture of oligodendrogliomas, when invading the cortex, neoplastic oligodendrocyte tend to cluster around neurons, a phenomenon referred to as „perineuronal satellitosis” (Micrograph 4). Accordingly, on the cut-surface, the cortical-subcortical border cannot be distinguished with naked eye, the border of the tumor tissue is blurred, however the frequently occurring multinodal calcification and intratumor haemorrhage are typical and prominent. Myxoid degeneration is in the background of the cut-surface sometimes being pulpy and shiny (Macrograph 4). This can be either due to the presence of neoplastic oligodendrocytes (used to be called mucocytes) accumulating a mucous PAS+ material, or due to the deposition of mucus-like material in the often microcystic intercellular matrix.
|
Look into the pictures!
|
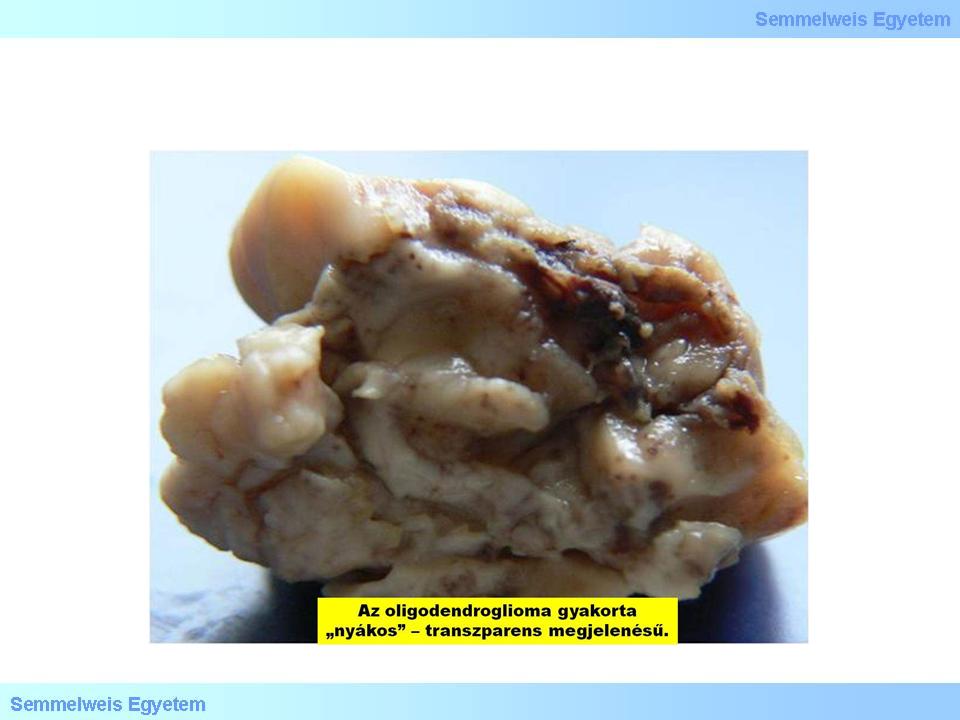
Macrograph 4: The appearance of an oligodendroglioma is often pulpy and shiny. (A favor of Péter Molnár; University of Debrecen, Medical and Health Sciences Center, Department of Pathology)
|
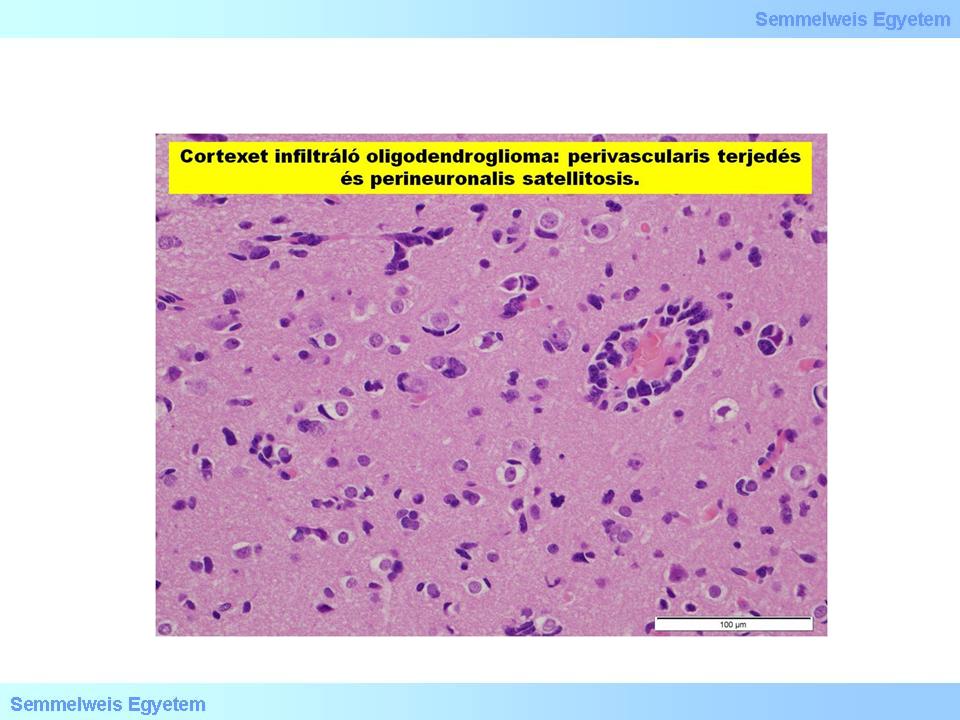
Micrograph 4: Oligodendroglioma invading the cortex: perivascular infiltration, perineuronal satellitosis. (A favor of Péter Molnár; University of Debrecen, Medical and Health Sciences Center, Department of Pathology)
|
The presence of „minigemistocytes” with intense eosinophilic cytoplasm, eccentric nucleus and smaller diameter compared to the gemistocyes described above, is of diagnostic value. The continuous reduction of differentiation (dedifferentiation) is inevitable in case of oligondrogliomas too: from the well differentiated GrII tumors to the definitely malignant and agressive, anaplastic GrIII forms. Nonetheless, glioblastoma multiforme does not develop from this tumor line. In the histologic picture of GrII tumors, vasculature of finely branching capillaries that may take on a “chicken wire” appearance, monomorphic nuclei, perinuclear halo (translucent cytoplasmic area around the nuclei of tumor cells) and extracellular calcification can be observed. (Note: The „halo” is a greek word referring to the circular track followed by the grain-treading horses, – this is also where the expression of halo/gloria above the head of saints, in painting, originates from!) Fifty percent of oligodendrogliomas are GFAP positive, due to the fact that at early developmental stages the neoplastic oligodendrocytes are capable of GFAP production.
II./2.3.5.2.: Anaplastic (malignant) oligodendrogliomas (GrIII)
|
|
The most important morphologic novelties are the necroses, the high nuclei-cell body ratio and the emerging cellular atypia that is accompanied by hypercellularity. At the same time, increased mitotic activity and vascular endothelial proliferation (MVP), as histopathologic signs of the agressively developing tumor, are also present. This tumor type tends to massively infiltrate the liquor spaces. Interestingly, this invasive cell clone is well differentiated and not anaplastic, however this process might soon result in a lethal outcome (hydrocephalus internus, herniatio tonsillarum cerebelli).
II./2.3.5.3.: Oligoastrocytomas
|
|
Mixed gliomas, oligoastrocytomas are very subjective and one of the most controversial categories of neuro-oncopathology. Two grades are known for these tumors (Gr.II, and GR.III). In Gr.III. oligoastrocytomas (Gr.III. OA) all histologic and cytologic characteristics of the glioblastoma can be found, but simultaneously, diffusely presenting, or clustering elements with perinuclear halo can also be observed. This criterion is highly dependent on the experience and subjective judgement of the evaluating pathologist. In order to avoid this subjective factor, a special category: „glioblastoma with oligodendroglial component” has started to be introduced. This issue, however, is about to remain without relevance along the results of large scale retrospective and prospective studies showing no impact of the presence of these mixed cell type composition on the patient survival rate (EORTC, 2012).
II./2.3.6.: Ependymoma (GrII)
The ependymoma macroscopically has generally well defined borders, it is rarely infiltrative. It is a soft, greyish-red and greyish-white, partially haemorrhagic, cystic, necrotic, slowly growing tumor, originating generally from the wall of the ventricules. The „classic” appearance is the most frequent in the region of the fourth ventricule, in the spinal cord, it is mostly an intramedullary mass. It has several histological types. In its histologic picture the perivascular pseudorosette is typical, that is tumor cells lined up around vessels with their processes direct toward the wall of the vessel, resulting in a perivascular empty area (without nuclei), which in turn causes finely fibrillar eosinophil spots in the orienting (low grade of magnification) microscopic view. This perivascular empty area is intensively GFAP positive. Low magnification in the microscope might be more useful in order to reach the proper diagnosis compared with strong magnification, because of the dominant presence of pseudorosettes.
|
Note
|
A further characteristic, and interesting histological sign is the true ependymal rosette. In these rosettes, and in the so called „ependymal canals”, elongated tumor cells are encircling a completely empty, central lumen (reminding to an embryonic medullary canal). These true rosettes are less frequent compared with pseudorosettes. Anaplastic (GrIII) form of ependymomas are also known, in which, next to pathocytologic and pathohistologic anaplastic signs, increased mitotic activity, pseudopalisading around necrotic tumor tissue, as well as vascular proliferation can be observed. Separately named histological type is the so called cellular ependymoma, characterized by an increased cellularity, that does not mean higher grade, by itself. Based on their immunoreactivity, ependymomas show GFAP-, vimentine-, nestin-, in certain cases S-100- and EMA-positivity, and in the anaplastic forms cytokeratin can also be detected. Rarely – similarly to other neuroectodermal tumors – melanin production also can occur. Specific forms are the tanycytic (Gr.II) and the myxopapillar ependymomas. The latter is mostly taken as Gr.I and mainly develops around the cauda equina. It grows slowly but in toto surgical removement is hardly possible in majority of the cases.
II./2.3.7.: Central neurocytoma (GrII)
Supratentorial, mainly intraventricular, gadolinum cumulating, frequently calcificated tumor that is isodens in CT. It can cause hydrocephalus by occluding the foramen Monroi. In intraoperative impression smear it can be observed that the cells are monomorphic, indeed look like oligodendroglia cells, but without processes. Besides the monomorphic cells, eosinophilic, nucleus free, fibrillar neuropil and partial rosette formation can be observed.
II./2.3.8.: Embryonic tumors
Their common feature is that their basic component are undifferentiated round cells, and as such, being part of the SBTC – small blue cell tumor group. The color refers to the deep basophilic feature of the cells with HE stain. In the microscope, at low magnification, the tumor tissue is deep blue, since the dominant component are the enlarged, hyperchrom nuclei, around which cytoplasma presents only as a narrow margin. Because of this, the whole tumor tissue consists of monotonic cell forms, that is called monomorphic atypia. Next to embryonic tumors (neuroblastoma, hepatoblastoma, pancreatoblastoma, nephroblastoma, etc.), microcytic lung cancer, small B-cell lymphoma and Ewing sarcoma are small blue cell tumors.
II./2.3.8.1.: Neuroblastoma (GrIV)
This is a highly malignant tumor typical in childhood. Its intracranial form typically develops supratentorially but it can also occur in the cerebellum. Additionally, it can also develop in extracerebral areas, practically anywhere in the peripheral sympathetic nervous system (mainly in the areas of paravertebral ganglions, adrenal medulla, and in paraganglional areas [e.g. organ of Zuckerkandl]). In classic neuroblastoma, neuroblasts and neuropil dominate (>50%), and neuroblast rosettes are frequent. Undifferentiated (macrocytic pleomorph form is also known), poorly differentiated and well differentiated types can be distinguished. Immunohistochemically the tumor tissue shows diffuse and strong NSE positivity. Other positive markers are: synaptophysin, NFP, ganglioside GD2, chromogranine. Proliferation index is, in general, between10-80%.
II./2.3.8.2.: Primitive neuroectodermal tumors (PNET, GrIV)
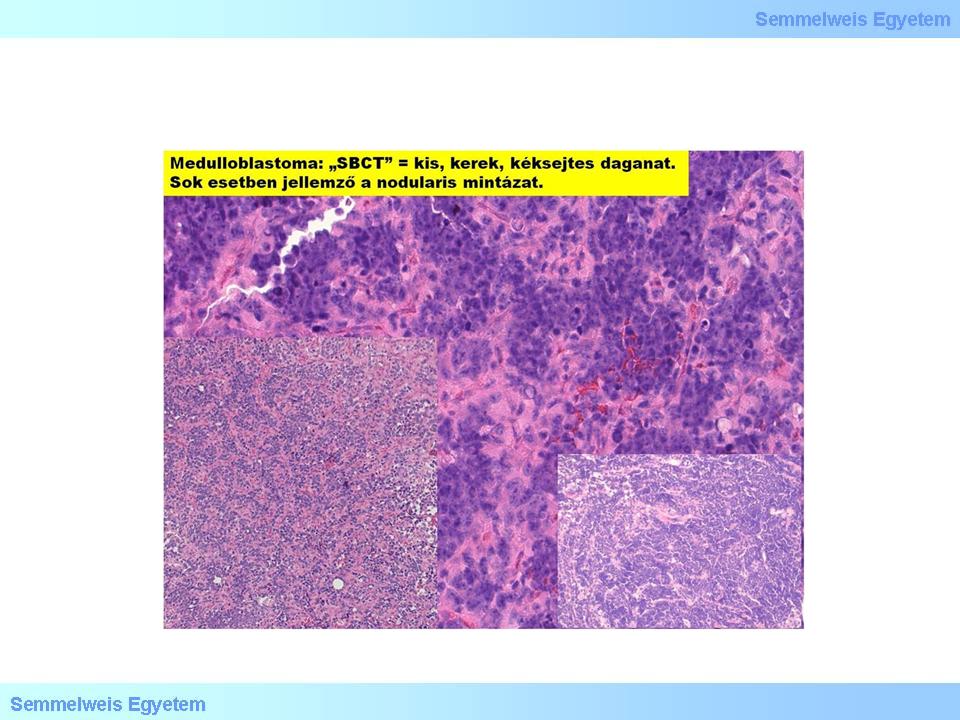
The PNET-concept is the most controversial topic of the field of classification of primary brain tumors. It is more frquent in the periphery but has already been described under different names supratentorially either: cerebral medulloblastoma, cerebral neuroblastoma, supratentorial SBCT (small blue-cell tumor) etc. The indeed various directions of development (neuronal, astrocytic, ependymal, muscular, melanotic), that takes place within a SBCT-environment, makes it a collective term. Histologically, the cells are anaplastic, sometimes indeed the SBCT characteristic is dominant, signs of differentiation cannot be observed. Nuclei are hyperchrome, the border of the small cytoplasma is blurred, the intercellular matrix is fibrillar but hardly detectable. The mitotic index is high (corresponding to Gr.IV). Bleeding and necrosis are not rare, occasionally, so called Homer-Wright rosettes occur (Hyperchrome tumorcells surrounding a lighter neuropil center). The primitive cells might express antigenes either typical for muscule tissue, or to glia cells. Supratentorial PNET cannot be distinguished from medulloblastoma cytologically, in the majority of cases.
II./2.3.8.3.: Medulloblastoma (GrIV)
It is typically a malignant, invasive, embryonic tumor of the cerebellum, the most prominent representative of the PNET-group. (The name is unfortunate, since „medulloblast” per se does not exist) This round tumor with slightly rough surface originates sometimes from the bottom of the fourth ventricule and fills in almost all of its lumen (Macrograph 5). It is histologically characterized by relatively definitive border, mainly small, anaplastic cells with hyperchrome nuclei and minimal cytoplasma, and with hardly detectable stroma. The presence of rosetta is not necessary for the diagnosis, it is often missing from these tumors. It is solid, homogeneusly cumulative, and leads rapidly to death without combined chemotherapy. It frequently, and extensively infiltrates the liquor spaces, and might give distant metastases along the neuroaxis. Since it is most frequently occurring in the posterior cranial pit, close to the ventricule, it frequently causes hydrocephalus. Immunihistochemically it shows a quite variable picture: it shows positivity to synaptophysine, neurofilament-proteins, nestin, vimentin, occasionally in star-like cells, to GFAP. The proliferation index is often >20%. The prognostic value of pathohistologic signs with regard to the clinical course and outcome is controversial; aneuploidy is a good sign.
|
Look into the macrograph!
|
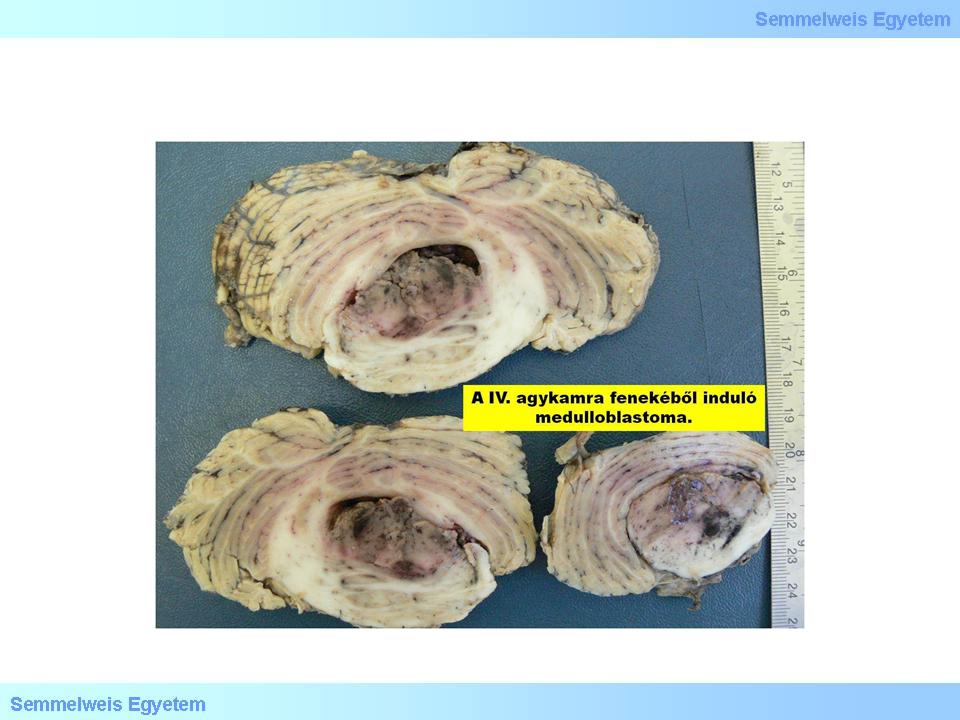
Macrograph 5: Medulloblastoma shows colorful appearance on the cut-surface. (A favor of Péter Molnár; University of Debrecen, Medical and Health Sciences Center, Department of Pathology)
|
Most recent research showed that PNET-medulloblastoma and the other SBCTs, although they have been considered uniform (micrograph 5), are, in fact genetically distinct. Untill the millenium, there had not been any reliable method either for the differentiation of these tumors, or for predicting their clinical outcome. However, applying the DNA-microarray technique, it was shown (2002), that the expression profile of medulloblastomas are different from that of supratentorial PNETs, and from that of other anaplastic, malignant gliomas. It was also proved that medulloblastomas originate from the stratum granulare cells, via the Sonic Hedgehog (SHH) regulating system. Recent studies also shed light to the role of SHH receptors (PTCH and GLI) and N-MYC in the development of desmoplastic medulloblastoma.
|
Look into the micrograph!
|
Micrograph 5: The medulloblastoma is a small blue cell tumor, which often shows a nodular pattern. (A favor of Péter Molnár; University of Debrecen, Medical and Health Sciences Center, Department of Pathology)
|
|