| |
III./3.: Komplexe kongenitale Herzfehler - DORV
Attila Tóth
III./3.1.: Einleitung
|
 |
Der Double outlet right ventricle (DORV, „doppelter Abgang des rechten Ventrikels“), wobei die großen Arterien Aorta und A. pulmonalis ausschließlich aus dem rechten Ventrikel entspringen, gehört zur Gruppe der angeborenen konotrunkalen Herzfehler. Erste Beschreibungen dieser Anomalität gehen zurück auf die Jahre 1793 und 1893. Spezifiziert wurde DORV dann schließlich 1952 durch Branu und seine Kollegen. Der Begriff „double outlet” geht nach Meinung diverser Autoren zurück auf AA Calhoun Witham.
Beim DORV handelt es sich um eine heterogene Anomalität, die sowohl eigenständig als auch im Zusammenhang mit anderen komplexen kongenitalen Herzfehlern auftreten kann. Je nach Fehlbildungskombination kann die Ausprägung dieses Herzfehlers und somit das Krankheitsbild sehr unterschiedlich ausfallen. So ist die klinische Manifestation z.B. abhängig von der Lokalisation des Ventrikelseptumdefekts (VSD) und dem möglichen Vorhandensein einer Transposition der großen Arterien. Charakteristisch für diese Anomalität sind ähnliche Druckverhältnisse in beiden Ventrikeln. Über den VSD, der eine interventrikuläre Kommunikation schafft und den einzigen Ausgang des linken Ventrikels darstellt, kann ein Blutabfluss über den rechten Ventrikel gewährleistet werden. Ohne operative Rekonstruktion kommt es aufgrund der erhöhten Druck- und Volumenbelastung zu einer Hypertrophie des rechten Ventrikels mit konsekutivem Hochdruck in der Lunge. Die so genannte Eisenmenger-Syndrome kann in dieser Konstellation nicht ausgelegt werden. Häufig geht die Anomalität mit einer Subpulmonalstenose (Bildnummer 5R-1.) einher, die in unbehandelten Fällen insofern hilfreich sein kann, dass sie die pulmonale Hypertension positiv beeinflusst. Durch eine begleitende Subpulmonalstenose und dem damit entstehendem Druckgradienten wird der Druck auf das pulmonale Gefäßsystem abgefangen und somit die Ausbildung einer pulmonalen Hypertension gemildert.
|
 |
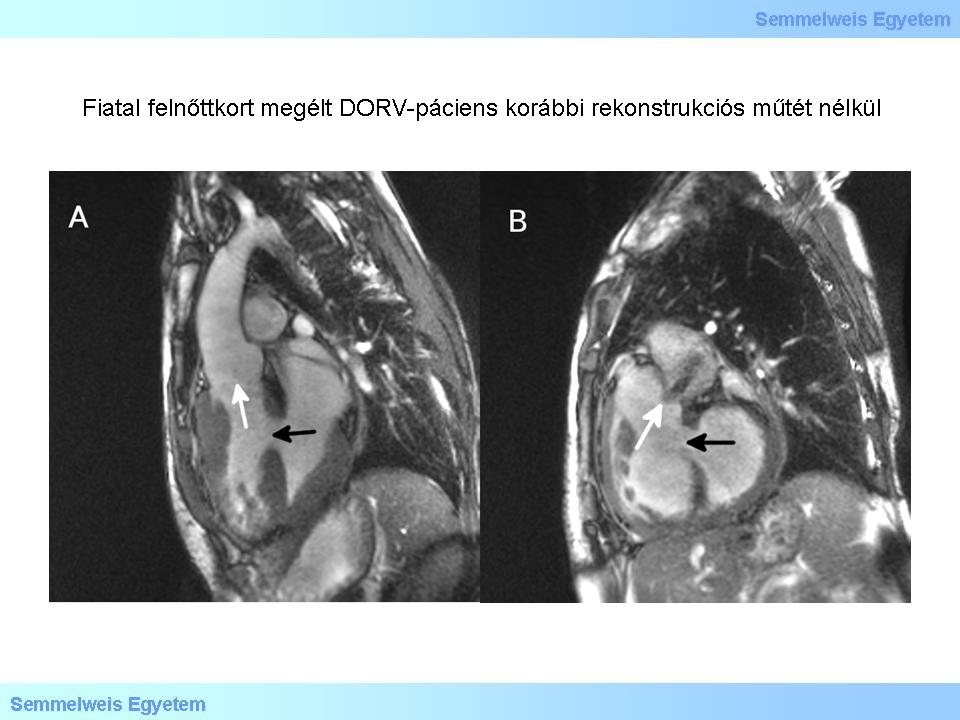
Abbildung 1: Patient mit DORV, der bis ins frühe Jugendalter ohne rekonstruktive OP auskam. A: Aortenwurzel. Schwarzer Pfeil – VSD; weißer Pfeil – Aortenwurzel aus dem rechten Ventrikel. B: Pulmonalwurzel. Schwarzer Pfeil – VSD; weißer Pfeil – Pulmonalwurzel aus dem rechten Ventrikel und anterograder Jet, der auf eine Stenose hinweist.
|
Mit einer Inzidenz von 0,033 - 0,145 pro 1000 Neugeborenen ist der DORV ein sehr seltenes Vitium. In unterschiedlicher Häufigkeit ist dieser Herzfehler mit anderen Fehlbildungen verbunden und es existieren verschiedene anatomisch oder funktionell orientierte Klassifikationen des DORV. Neben dem immer vorkommenden VSD werden parallel auftretende Pulmonalstenosen, AV-Klappen-Anomalitäten, Stenosen des rechten Ausflusstrakts (RVOT, right ventricular outflow tract) und obstruktive Anomalitäten des Aortenbogens häufig beobachtet. In selteneren Fällen treten darüber hinaus auch atrioventrikuläre Septumdefekte (AVSD), Juxtapositionen des linken Herzohres, supero-inferiore Ventrikel, gekreuzte atrioventrikuläre Konnektionen, kongenitale Defekte des systemischen oder venösen pulmonalen Rückstroms oder Ventrikelhypoplasien auf.
Bei einer Spezialform des DORV, dem Taussing-Bing-Syndrom, liegt eine Transposition der großen Gefäße vor. Der Kammerscheidewanddefekt ist in diesem Fall unterhalb der Lungenarterie lokalisiert (Bildnummer 5R-2.) (Benannt wurde diese Anomalität nach Helen B. Taussig und Richard J. Bing, die die Therapiestrategien dieser kongenitalen Herzfehler entscheidend geprägt haben). Stenosen des rechten Ausflusstrakts gehen oftmals mit Anomalitäten der AV-Klappen und einer Aortenisthmusstenose einher. Ziel des chirurgischen Eingriffs ist eine biventrikuläre Korrektur, wobei häufig das Prinzip der Rastelli-Operation zum Einsatz kommt. Hierbei werden die Kreisläufe auf der Ebene der Ventrikel durch das Einsetzen eines klappentragenden Patchs, der einen funktionellen Verschluss des VSD zu Folge hat, voneinander getrennt; die Kontinuität zwischen rechtem Ventrikel und Pulmonalarterie wird über ein Patch hergestellt. Hierbei handelt es sich in den meisten Fällen um ein Homograft, also um ein Gefäß humanen Ursprungs, das in Gewebebanken spezialisierter Institutionen für Patienten asserviert wird.
|
|
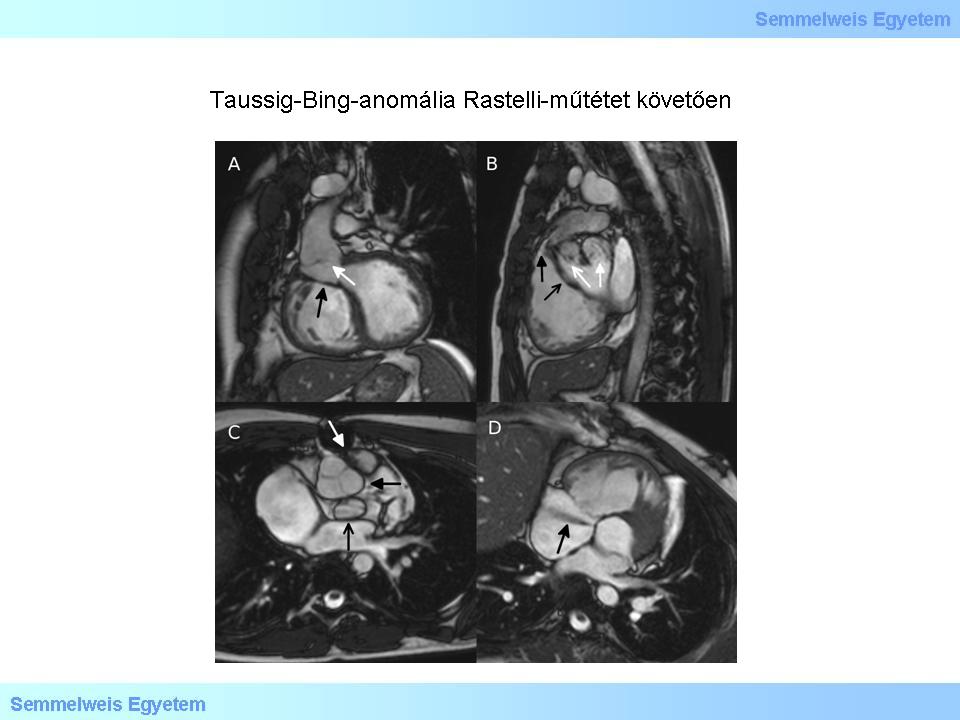
Abbildung 2: Taussig-Bing Anomalität; Indikation zur Rastelli-OP. A: Ausfluss des rekonstruierten linken Ventrikels. Weißer Pfeil – Blutfluss über den VSD in die Aorta entlang des Patchs; schwarzer Pfeil – Patch. B: Homograft, der im Ausflusstrakt des rechten Ventrikels seinen Ursprung hat. Dicker schwarzer Pfeil - anterograder Jet, der auf eine Klappenstenose hinweist. Dünner schwarzer Pfeil –Patch; dicker weißer Pfeil – Überbleibsel der ursprünglichen Pulmonalarterie, der keine Funktion mehr zukommt; dünner weißer Pfeil – Aortenursprung. C: Wurzeln. Dicker weißer Pfeil – Ursprung des Homografts; dicker schwarzer Pfeil – Aortenklappe mit drei Taschen; dünner schwarzer Pfeil - bikuspide Pulmonalklappe (außer Funktion). D: Shunt vom Gerbode-Typ. Schwarzer Pfeil – Gerbode-Jet.
|
Das Kapitel Struktur
|
|
Referenzen
|
 |
Freedom RM et al: The Natural and Modified History of Congenital Heart Disease. 19A: The Divided Right Ventricle. Wiley-Blackwell, 2006, pp. 232-235.
Freedom RM et al: The Natural and Modified History of Congenital Heart Disease, 28: Double-Outlet Ventricle. Wiley-Blackwell, 2006, pp. 370-380.
|
|