| |
II./2.3.: Primär parenchymatöse Tumore des Gehirns
II./2.3.1.: Primärtumore des ZNS
|
 |
Am Präparat kann man sehr guter eine der wichtigsten makroskopisch sichtbaren Eigenschaften eines des häufigsten Vertreters der intrakranialen (ic) raumfordernden Prozesse demonstrieren: es handelt sich hier um Neoplasien, um primäre Gehirntumoren. Die intrakranial vorkommenden Tumore können in zwei grosse Gruppen eingeteilt werden (was auch für die Tumore des Rückenmarks gilt): es kann sich um primär aus neuronalen Zellen gebildete Tumore, oder von anderswo hierher eingeschwämmte, metastatische (sekundäre) Tumore handeln. Die malignen Tumore, die aus den das Gehirn umgebenden Geweben in das Gehirn einwachsen, werden an anderer Stelle besprochen.
Die Inzidenz der primären Hirntumore beträgt 7-10/100.000 Einwohner. Diese Tumore kommen leider besonders häufig im Kindesalter vor (sie stehen an 2. Stelle der am häufigsten im Kindesalter vorkommenden Tumore); ein zweiter Peak zeigt sich im Alter von 45 bis 70 Jahren (2D). Die Ätiologie dieser Tumore ist meist unbekannt, obwohl bei einigen Tumoren eine familiäre Anhäufung (sog. Tumor-Syndrome), schädigende Umwelteinflüsse (meningeale Tumore bilden sich oft nach einer Strahlenbelastung der Kopf- Halsregion), oder ein inadäquat funktionierendes Immunsystems (Zusammenhang zwischen einem primär zerebralen Lymphom (meist aus B-Zellen) und AIDS).
II./2.3.2.: Einteilung der primären Gehirntumore
|
 |
Die meisten primären Hirntumore sind neuroektodermaler Herkunft. Zu dieser Gruppe gehören ferner noch die Lymphome des zentralen Nervensystem (CNS - central nervous system), die Tumore der Gehirnhäute (Dura Mater, Arachnoidea), usw. mit denen wir uns hier aber nicht beschäftigen. Die neuroektodermalen Tumore sind als erstes von Percival BAILEY und Harvey CUSHING eingeteilt worden (1926). Diese beiden Forscher teilten die Tumore nach ihren zyto- bzw. histogenetischen Gegebenheiten ein. Sie bauten ihre Theorie auf den Gedanken auf, dass die Strukturen der Tumore - abhängig von ihrem Differenziertheitsgrad - den Zellen ähneln, von denen sie abstammen. So war es vollkommen logisch, dass die wichtigsten Tumore in zwei grosse Gruppen eingeteilt worden sind:
-
- aus der Glia stammende Tumore (Gliome): Astrozytome, Oligodendrogliome, Ependymome; bzw
-
- aus den Nervenzellen stammende Tumore ( "Ganglien-Zell" Tumore).
Die Zellen eines Tumors, die sich in der frühen Entwicklungsphase befinden (Spongioblast, Medulloblast, Astroblast,usw.), hatten den Forschern nach ein besondere zytogenetische Bedeutung - obwohl die Forschung damals diese Bezeichnung noch nicht benutzt hat. Durch die Weiterentwicklung der neurochirurgischen Techniken konnten immer mehr Tumortypen differenziert werden, und es stellte sich heraus, dass die Tumore oft nicht nur aus einer Zellart bestehen, sondern dass sich in ein und dem selben Tumor verschieden ausgebildete Zellelemente befinden können ( gemischtzellige Gliome ( z.B.Oligoastrozytom), neuronale Gliazell-Tumore (z.B. Gangliogliome)).Die Einteilung hat sich schliesslich über einen langen, komplizierten Weg weiterentwickelt, dessen Einzelheiten nur noch für die Geschichte der Medizin von Bedeutung sind.
|
 |
Die heute offizielle akzeptierte Variante der morphologischen Klassifikation (morphe - Äusseres; morphologisch- dem Aussehen entsprechend) ist die WHO Einteilung von 2007, die mehr als 100 Tumortypen, bzw. Tumor Untertypen voneinander unterscheidet. Die histologische Einteilung der Tumore nach Kernohan brachte eine geringgradige Vereinfachung der sich schnell entwickelnden histologischen Einteilung dieser Tumore. Kernohan arbeitete an der Mayo Klinik (Rochester, MN, USA), und übernahm sehr früh schon die von seinem Kollegen Albert Broders 1920 veröffentlichte Ansicht, wonach man das klinische Verhalten eines Tumors (sein biologisches Potential) am einfachsten mit Hilfe einer numerischen Reihe einordnen kann.
Broders teilte die Karzinome der Mundschleimhaut in vier verschiedene Gruppen (Grade) ein.
-
- In solche, bei denen über 75 % der Tumorzellen dem Ausgangsepithel ähneln (d.h., wenn der Tumor hoch differenziert ist); in diesem Fall wurde der Tumortyp als Grad I bewertet;
-
- ähnelten 50-75% der Zellen dem des Ausgangsepithels, dann wurde der Tumor zum Grad II eingeteilt (Gr.II);
-
- betrug der Prozentsatz der differenzierten Zellen nur noch 20-25%, so wurde der Tumor als Typ III bezeichnet (Gr.III)
-
- und die kaum differenzierten Tumore (die also ausgesprochen anaplastischen Tumore), bei denen weniger als 25% der Zellen denen des Ausgangsepithels ähnelten, wurden in die Gruppe der Gr IV Tumore eingeteilt.
Kernohans Vorstellung, dass dieses Einteilungssystem auf die Tumore des ZNS adaptierbar ist, bewies sich leider als falsch. Die Aufteilung in die 4 Gruppen der sich verschieden differenziert verhaltenden Tumore konnte - mit Einwänden - noch am ehesten bei den astrozytären Tumoren angewandt werden (aber auch nicht ohne Vorbehalte); bei den Oligodendrogliome und den Ependymomen gab es nur Tumore des Grades II und III. Die eigentliche Vorstellung, die als Grundlage dieser Einteilung diente, hat sich auch um einiges geändert, da heutzutage auch solche Zelltypen (Phänotyp) bei der Bestimmung des Tumorgrades ausschlaggebend sind, die im Nervensystem eigentlich nicht vorkommen (s.auch "Pilozyt" - pilozytäres Astrozytom). Im Gegensatz zur eigentlichen Vorstellung von Kernohan dient heutzutage bei der histologischen Beurteilung eines Tumors nicht nur die Ähnlichkeit der Tumorzellen umschriebenen" (Gr I.) Tumore - Astrozytom, Pilozytom - und die "diffusen" Tumore, die keine scharfe Grenze haben - also eigentlich alle anderen Gliome - in der WHO Einteilung von 2000 das erste Mal als eigenständige Gruppe aufgeführt worden, was aus biologisch-klinischer Sicht eine wichtige Bedeutung hatte. Ebenfalls zu beachten ist, dass bei der Einteilung der verschiedenen Tumorgruppen deren biologisches Verhalten (Grad) auch weiterhin eine Rolle bei der Einstufung spielt. All dies kann am besten am Beispiel der astrozytären Gliome beobachtet werden (Tabelle 1).
|
Beurteilen Sie die Tabelle!
|
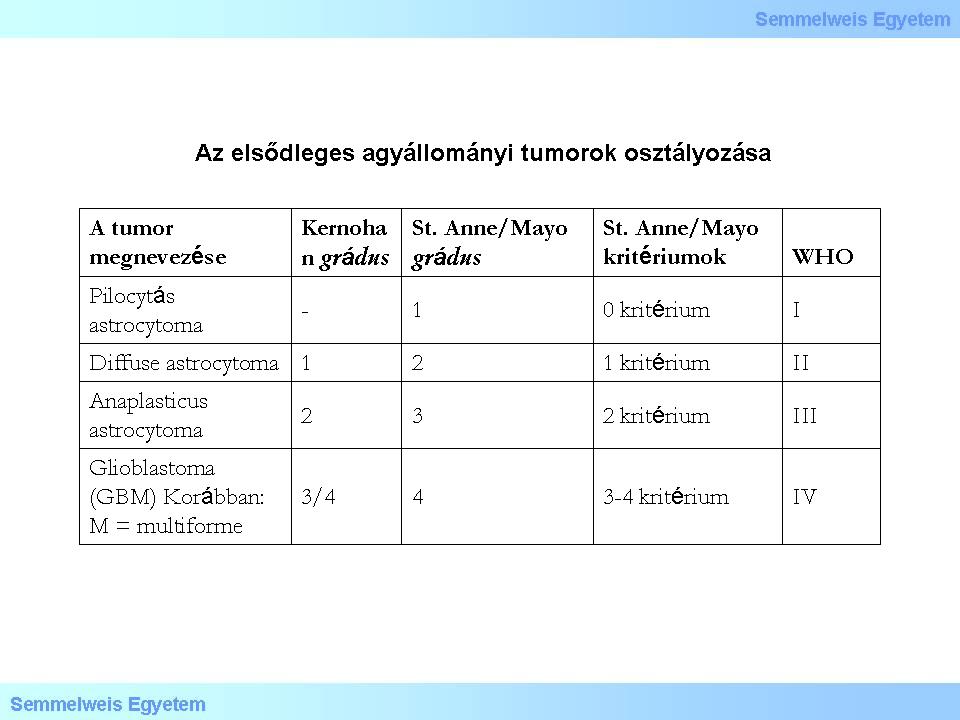
Tabelle 1.
|
Die Prognose eines primären Hirntumors ist in erster Linie von der Zytologie des Tumors, bzw. den molekulären Abnormalitäten vor/während der Tumorbildung abhängig. Am wichtigsten hierfür ist die "Zellreife" des Tumors (sog. zytologische Differenzierung), die auch schon am längsten benutzt wird. In den vergangen 5-10 Jahren ist die Bedeutung der molekularen/genetischen Schäden und die zytogenetischen Veränderungen immer mehr in den Vordergrund gerückt. Die bis ins Extreme gehende Vielfalt der Zellpopulationen innerhalb eines Tumors (Heterogenität) erschwert sehr oft die Beurteilung des Differenzierungsgrades (die Reife) eines Tumors - den sog. Grad des Tumors (auch das englische Wort `grade` stammt aus dem selben lateinischen Wort, das so viel wie "Treppe" bedeutet). So kann es z.B. sehr oft vorkommen, dass ein am Randgebiet des Gehirns reif erscheinendes Gliom in den tieferen Regionen, die schwerer erreichbar sind, anaplastische Tumorzellen beinhaltet, also ein Glioblastom (GBM) darstellt. Die Einteilung der Tumore geschieht trotz der Schwierigkeiten aufgrund des Gewebsaufbaus, weil dieser noch am ehesten proportional zur Prognose des Tumors ist. So müssen also zur Bestimmung des Reifegrades z.B. eines Astrozytoms, das den best erforschten Tumor darstellt, 4 bestimmte Kriterien - im übertragenen und wörtlichen Sinne- "unter die Lupe genommen" werden:
-
1) Atypien des Zellkerns,
-
2) zytologische Pleomorphie,
-
3) Zellteilungsaktivität,
-
4) eine Endothelzellhyperplasie und/oder eine Nekrose, die von einem Tumorzellkranz umgeben wird (die Tumorzellen stellen sich zaunartig auf (meist werden so die Nekrosen umzäunt), und bilden so eine Pseudopallisade).
Dem zufolge können 4 verschiedene Reifegrade/Differenzierungsgrade (grade) unterschieden werden:
-
- GrI (grade I) - keine der beschriebenen Veränderungen ist im Tumor zu sehen;
-
- GrII - eine der 4 Veränderungen ist im Tumor enthalten;
-
- GrIII - 2 von 4 Veränderungen sind zu sehen;
-
- GrIV - 3 der oben beschriebenen Veränderungen sind im Tumor enthalten.
Diese Aufteilung diente als Grundlage für die sog. Saint Anne-Mayo Klassifikation. Danach kamen die Einteilungen der WHO, die immer wieder etwas an der ersten Einteilung änderten, die Grundprinzipien wurden aber immer akzeptiert, und sind mehr oder weniger beibehalten worden. Das biologische Verhalten der Gliazelltumore wird von ihrem starken Hang zur Infiltration bestimmt. Heutzutage kann ein Grade I -Tumor dieser Gruppe eigentlich nicht mehr diagnostisiert werden; für diese Form des Tumors ist ein umschriebenes ("expansives") Wachstum typisch. Im Gegensatz dazu zeigt jeder Tumor höheren Grades dieser Gruppe ein diffuses Wachstum.
Ein weiteres wichtiges Kriterium, das den klinischen Verlauf eines Gehirntumors beeinflusst, ist seine anatomische Lokalisation. Diese Tatsache kann bei einigen Tumoren sogar als diagnostisches Kriterium dienen, da diese sich immer an der selben Stelle bilden (Prädilektionsstellen bestimmter Lokalisation). Ferner hat man festgestellt, dass die Lokalisation eines Gehirntumors seinen sogenannter molekularer "Fingerabdruck" beeinflusst: ein sich im Frontallappen bildendes GBM zeigt oft ganz andere Charakteristika als der selbe Phänotyp des GBM, das sich im Okzipitallappen ausbildet! Wegen eben diesen Zuständen können oft sogar histologisch "benigne, differenzierte" Tumore (mit einer guten Prognose) innerhalb kürzester Zeit zum Tode des Patienten führen (z.B. ein Meningiom der hinteren Schädelgrube, oder ein sich im IV. Ventrikel bildendes, gut differenziertes Ependymom, usw.). Jegliches Tumorwachstum stellt einen raumfordernden Prozess dar, und hat den Anstieg des intrakraniellen Druckes zur Folge.
Im Laufe der Zeit kommt es mit dem Fortschreiten des Tumorwachstums fast immer zu seiner Dedifferenziation. Trotz alldem zeigen auch dedifferenzierte intrakraniale Tumore kaum den Hang zur extrakranialen Metastasierung (8B); ihr biologisch aggressives Verhalten verbirgt sich in ihrem starken Hang zur Rezidivbildung. Die Einreihung der primären Gehirntumore geschieht heutzutage nach der WHO Aufteilung von 2007, obwohl auch diese Einteilung heute eigentlich schon als veraltet gilt.
II./2.3.3.: Onkogenese der primären Hirntumore
|
 |
Die Benennung der einzelnen Tumore erweckt oft den Eindruck , dass sich ein Tumor aus einer endgültig definierten Zellart bildet. Ein typisches Beispiel hierfür ist die Benennung des "Oligodendroglioms". Dies wirft einige theoretisch Fragen, vor allem aber die auf, ob dieser Tumor auch wirklich aus Oligodendrozyten besteht. Man hat laut den neuesten Forschungen feststellen können, dass das vollständig ausgebildete menschliche Gehirn an einigen Gebieten zu einer Zellteilung fähig ist, und man dort pluripotente Progenitorzellen finden kann. Einige dieser Zellen spielen ganz sicher eine Rolle bei der Tumorgenese der glialen, bzw. neuroektodermalen Tumorgenese. Man ist sich heutzutage auch sicher, dass die Schädigung der DNS zu bisher für unmöglich gehaltenen Veränderungen auf Zellebene fähig ist: es kann sowohl zu einer mesenchymal- epithelialen als auch zu einer epithelial- mesenchymalen Transformation (MET und EMT) kommen; es kann also zu einer sog. regressiven Differenziation, bzw. einer retrograden Veränderung von ausgebildeten Zellen kommen, in Anbetracht dessen sogar nicht einmal die bizarre Vorstellung ausgeschlossen werden kann, dass Oligodendrozyten nach einer genetischen Schädigung fáhig sind, sich dermassen zu dedifferenzieren, dass sich ein Tumor bildet. Anhand der folgenden, relativ neuen (2010), aus einer Publikation übernommenen Abbildung (Abb.1) kann man sehen, wie man sich heute die Bildung eines Glioms vorstellt. Die gestrichelten Pfeile zeigen, dass es aus dieser Hinsicht auch heute noch viele offenen Fragen gibt. Es ist wichtig zu bemerken, dass es auf dieser Abbildung keinen möglichen Weg für die Ausbildung eines Ependymoms gibt!
|
Beurteilen Sie die Abbildung!
|
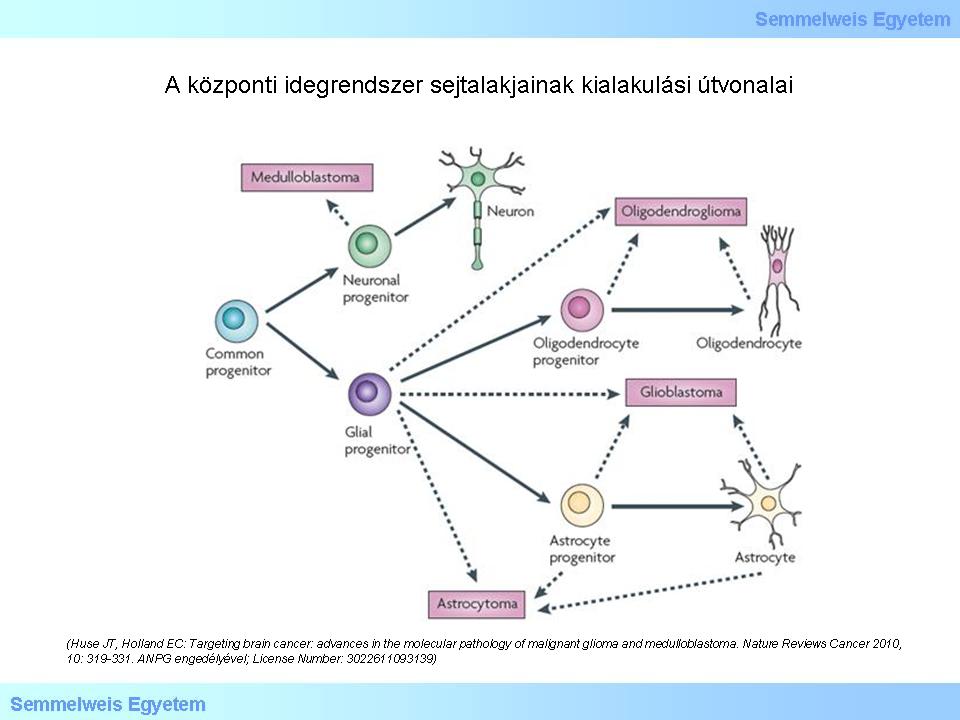
Abbildung 1: Bildung der Zellen des zentralen Nervensystems (central nervous system, CNS)(JT Huse, Holland EC: Targeting brain cancer: advances in the molecular pathology of malignant glioma and medulloblastoma. Nature Reviews Cancer 2010, 10...)
|
Dieser Tabelle zufolge stammen die in verschiedene Richtungen differenzierten Zellen von einer gemeinsamen Progenitorzelle ab. Kommt es während der Differentiation einer bestimmten Zelle aus dieser pluripotenten Vorform ( die eigentlich keine wirkliche "Stammzelle" darstellt) zu einer genetischen Störung, kann sich daraus ein ungebremstes Zellwachstum („immortalisatio”) bilden, was zur Ausbildung eines Tumors führt. Die modernen Möglichkeiten, die es inzwischen zur Untersuchung des Genoms gibt, haben es möglich gemacht, den molekularen Mechanismus, der für die Ausbildung der meisten Gliome verantwortlich ist, zu beschreiben. Das Genom des GBM ist in einem Artikel von 2008 erstmals beschrieben worden. Es hat sich herausgestellt, dass die Mutationen verschiedener Rezeptor Kinasen (RTKs), so wie auch z.B. der epitheliale Wachstumsfaktor (EGFR) und der Thrombozyten abhängige Wachstumsfaktor (PDGFR), eine wichtige Rolle bei der Genese der Gliome spielen. Die Wachstumsfaktoren üben ihre onkogenetische Wirkung durch die Signaltransduktion des PI3K-AKT- m TOR und des RAS-MAPK aus. Aus dieser Tabelle ist weiter ersichtlich, dass die Defekte des p53 und des RB Suppressors ebenso ausschlaggebend bei der Tumorigenese sind (9B). Die Schädigung dieser Gene führt zur Bildung abnormaler Proteine. Diese abnormalen Proteine ermöglichen dann die Unterscheidung der einzelnen Typen eines Glioms (in erster Linie die der Glioblastome). In der 2. Abbildung ist der EGFR rot, der PDGFR blau und der NF1 Rezeptor mit einer grünen Farbe gekennzeichnet. Ferner kann man sehen, dass in der Tumorgenese die aus der allgemeinen Onkologie und den Zellteilungszyklen bekannten wichtigen Moleküle hier ebenfalls eine wichtige Rolle spielen.
|
Beurteilen Sie die Abbildung!
|
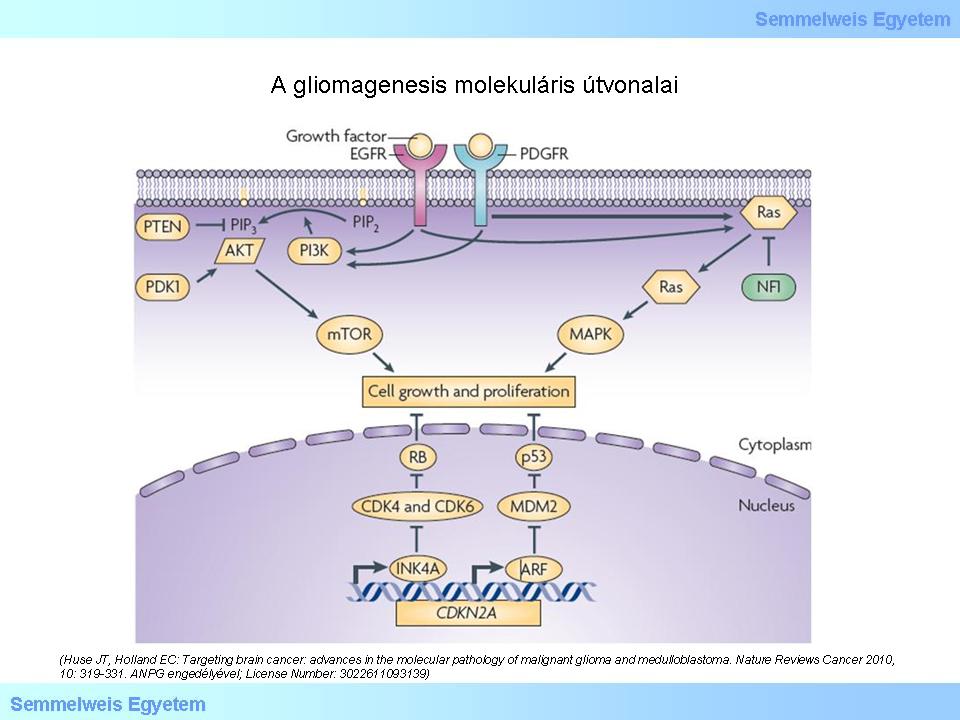
Abbildung 2: Molekuläre Kaskade der Gliomgenese. CDK : Zyklin-abhängige Kinase. NF1 : Neurofibromin. PDK1 : 3-Phosphoindosit-abhängige Proteinkinase. PIP2 : Phosphatidylinositol -4,5- Bisphosphat. PIP3 : Phosphatidylinositol -3,4,5- trisphosphat (JT Huse, Holland EC: Targeting brain cancer...)
|
Im "blauen Buch" der WHO 2007 wurde bereits auf die Wichtigkeit dieser Tumorbildungskaskaden hingewiesen. Die schematische Abbildung sieht man weiter unten (Abb.3), aus der auch klar ersichtlich ist, dass die klassische "genetisch/biologische Progression" (Gr II.- Gr III.- Gr. IV.) eine der Kaskaden darstellt, aus der sich die sekundären Glioblastome bilden. Die Gruppe der primären GBM zeigt eine vollkommen andere Bildungskaskade.
|
Beurteilen Sie die Abbildung!
|
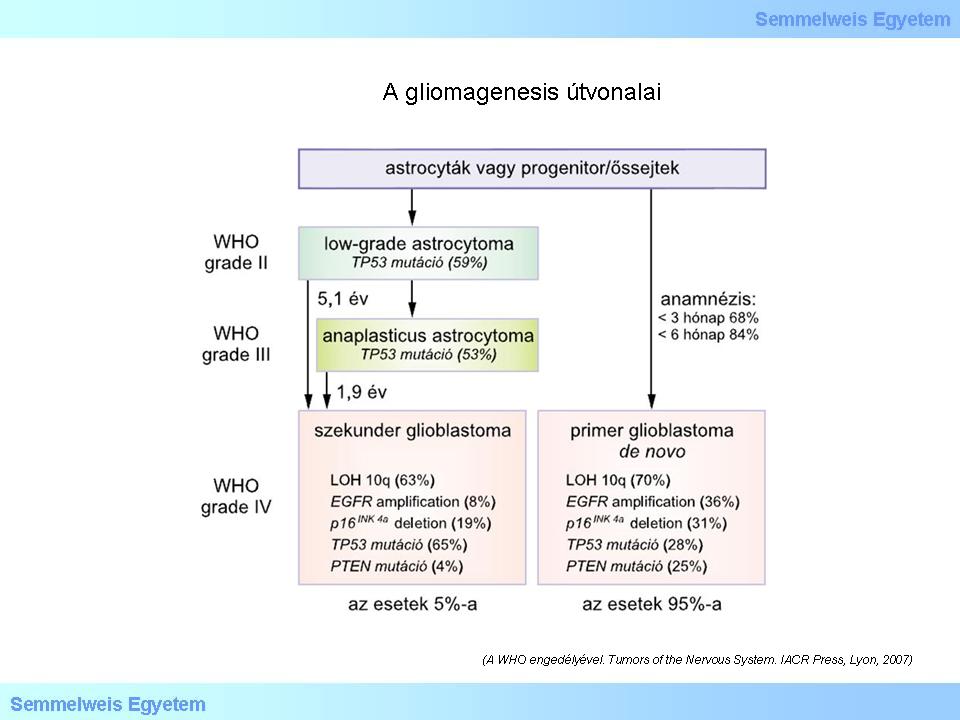
Abbildung 3: Bildungskaskade eines Glioms (mit Erlaubnis der WHO. Tumors of the Nervous System. IACR Press, Lyon, 2007).
|
Bei der Erforschung der Tumorigenese (genauer, s.: Miklos Kásler, Lehrbuch der klinischen Onkologie) ist - sozusagen als Nebenbefund - erkannt worden, dass in den meisten Gliomen der erste Genfehler in einem metabolischen Enzym (Isozitratdehydrogenase -1: IDH -1) erscheint. Das IDH -1 ist ein zytoplasmatisches Enzym, bei dem die Missense- Punktmutation an Stelle R 132 (aktives Zentrum des Enzyms) lokalisiert ist. Heutzutage weiss man auch, dass eine weitere Mutation an Stelle R172 des mitochondrialen IDH -2 Enzyms, zwar seltener, aber in Form einer Punktmutation ebenfalls vorkommen kann. Die IDH -1/IDH- 2 Mutation kommt in erster Linie in Gliomen niedrigen Grades in der frühen Phase der Tumorbildung vor. Wird dieser Fehler von einer Mutation am 1p19q -Lokus, einer Deletion/ko-Deletion gefolgt, dann verhält sich der Tumor einem Oligodendrogliom entsprechend, und sieht auch oft dem entsprechend aus. Folgt als zweiter Schritt eine Mutation des p53, bildet sich ein anaplastisches (Gr III.) Astrozytom, und später ein sekundäres Glioblastom (Gr IV. GBM). Die Mutation des IDH ist deshalb so ausschlaggebend, weil dieses zytoplasmatische Enzym bei der Lipidsynthese, dem Schutzmechanismus gegen oxidativen Stress, bei der oxidativen Zellatmung und bei der Sauerstoff- Sensor Signaltransduktion eine Schlüsselrolle spielt.
|
|
Bei der Frage, durch welchen Mechanismus die Metabolite, die bei der Funktion des IDH -1 katalysiert werden (2HG=2 -Hydroxiglutarat), auf die Bildung eines Gehirntumors und auf die der akuten Leukämie im Erwachsenenalter wirken, scheiden sich die Geister. Heutzutage wird ohne immunhistochemische Untersuchungen des mutierten IDH -1 Proteins keine Diagnosestellung vorgenommen; sind im Tumor Zellen zu sehen, die morphologisch einem Oligodendrogliom entsprechen, ist auch die Untersuchung des 1p19q mit einer FISH ein Muss. Die Grad IV. Gliome (Glioblastome) werden heutzutage, aufgrund von genetischen Untersuchungen, in 4 Untertypen aufgeteilt. Diese Aufteilung hat eine wichtige prognostische Bedeutung hinsichtlich der Überlebensaussichten des Patienten. Der Grad der Methylation am Promotor des 1p19q LOH Enzyms, und an dem des MGMT (Methylguanin- Methyltransferase) stellt wirklich einen "prediktiven" Marker dar. Letztere zeigen, auf welche Medikation ein Tumor gut reagieren wird (beim 1p19q LOH wirkt PCV, bei der Methylation des MGMT sollte man Temodal geben). Die Besprechung aller Einzelheiten dieses Themas würde den Rahmen dieses Kapitels sprengen (Tabelle 2).
|
Beurteilen Sie das Bild!
|

Tabelle 2: Man unterteilt die malignen Gliome heutzutage in 4 Untergruppen: (1) proneural; (2) mesenchymal; (3) klassisch; (4) neural. Die molekulare Pathogenese und der klinische Ablauf dieser Untergruppen unterscheiden sich voneinander. Die Erforschung der Einzelheiten dieser Tumore stellt eines der wichtigsten Themen der modernen Neuro- Onkologie dar (JT Huse, Holland EC: Targeting brain cancer: ....).
|
II./2.3.4.: Astrozytome
II./2.3.4.1.: Allgemeine Charakteristika der Astrozytome
Das Astrozytom kann in jedem Lebensalter und jeder Lokalisation vorkommen. Meistens wächst der Tumor infiltrativ, weshalb man ihn oft nicht vollständig (in toto) entfernen kann. An den Tumorrändern bildet sich eine reaktive Astrozytose, die auch mit den modernsten diagnostischen Verfahren nur sehr schwer von Ausläufern des Astrozytoms unterschieden werden können, was heisst, dass neoplastische Teile des Tumors kaum von reaktiven Gebieten zu unterscheiden sind. In solch einem Falle kann eine immunhistochemische Reaktion mit IDH -1 (s.oben) hilfreich sein. Intraoperativ sollte deshalb eine Fluoreszenz Färbung vorgenommen werden, damit tumoröse von normalen Gebieten unterschieden werden können. Die Behandlung solch eines Tumors ist deshalb sehr schwer, weil vor allem im Areal um einen gut differenzierten Tumor die Blut-Hirn Schranke nur moduliert funktioniert, die wasserlöslichen chemotherapeutischen Stoffe die Blut-Hirn Schranke nicht überschreiten können, und so den Tumor nicht erreichen. Die aus der Glia stammenden Tumore sind leider auch oft strahlenresistent. Dies bedeutet also, dass der Tumor weder chirurgisch komplett entfernt werden kann, noch die Chemo- und Radiotherapie den Tumor vollständig eradiieren können, was diesem Tumor so eine schlechte Prognose verleiht: bei Patienten, die an der agressivsten Form eines diffusen malignen Glioms - an einem Glioblastom - leiden (was leider sehr oft vorkommt), beträgt die durchschnittlicher Überlebensdauer, trotz moderner Behandlungsmethoden, immer noch lediglich 12 - 14 Monate. Aus dieser Hinsicht hat die rapide Erforschung des molekularen "Fingerabdruckes" dieser Gliome einen Durchbruch ergeben: ist bei einem betroffenen Patienten gleichzeitig sowohl die IDH -1/ IDH -2 Mutation, als auch die Methylierung des Promotors beim NGNT -Enzym vorhanden, kann man bei der Gabe einer kombinierten Chemo- Radiotherapie (sog. Stupp Protokoll) immer öfter eine Überlebensdauer der Patienten von bis zu 2-4 Jahren erreichen!
II./2.3.4.2.: Hoch differenzierte Astrozytome (Gr I.)
|
|
Oft benötigt diese Art von Tumor keinerlei radikale Behandlung. Die hochdifferenzierten Astrozytome wachsen langsam, und sind je nach Lokalisation lange Zeit symptomlos. Zur Gruppe dieser Tumore gehören die vom N.opticus und vom Chiasma Opticum ausgehenden astrozytären Gliome, und das zerebellare Astrozytom. Zur Diagnosestellung nimmt man aus diesem Tumor eine mit Ultraschall oder einem CT kontrollierte, stereotaktische Biopsie. Die Grad I Astrozytome werden auch pilozytäre Tumore genannt. Das zerebelläre pilozytäre Astrozytom stellt logischerweise einen zystischer Tumor dar - was auch mit bildgebenden Verfahren nachgewiesen werden kann -, bei dem sich die Tumormasse in die Zyste hineinstülpt (was so eine Art "Knoten" entstehen lässt). Bei der Betrachtung des zytologischen Abdruckes dieses Tumors zeigen sich relativ monomorphe, längliche, spindelförmige, bipolare Zellen mit langen Ausläufern, und runden, bzw. eierförmigen Zellkernen, die ein gekörntes Chromatin enthalten. Es ist keine wirkliche Zell- bzw. Kernpolymorphie zu sehen. Ein Teil dieser Tumore kann chirurgisch entfernt werden, was eine sehr gute Prognose bedeutet. (Bemerkung: Im Wegweiser der WHO 2007 sind die erlaubten morphologischen Abnormitäten der pilozytären Astrozytome so aufgezählt, dass sie leicht mit den Charakteristika eines GBM verwechselt werden können - eine Diagnosestellung ist längst nicht so einfach, wie man anhand der didaktisch vereinfachten Beschreibung denken könnte!)
II./2.3.4.3.: Diffuse Astrozytome (gut differenziert; Gr II.)
In dieser Gruppe der Tumore (15P-1. Makrofoto; 15P-1.Mikrofoto) unterscheidet man histologisch eine fibrilläre, protoplastische und gemistozytäre Form. Eine wichtige onkologische Charaktere dieser Tumore ist ihr Hang zur Progression (stetige Minderung der Ausreifung, Dedifferenzierung). Diese Progression im klinischen/biologischen/molekulären Sinne, also die andauernde Akkumulation der genetischen Fehler, ist nicht mit der radiologischen Progression, also der Vergrösserung bzw. dem Wachstum des Tumors zu verwechseln!
|
Beurteilen Sie das Bild!
|
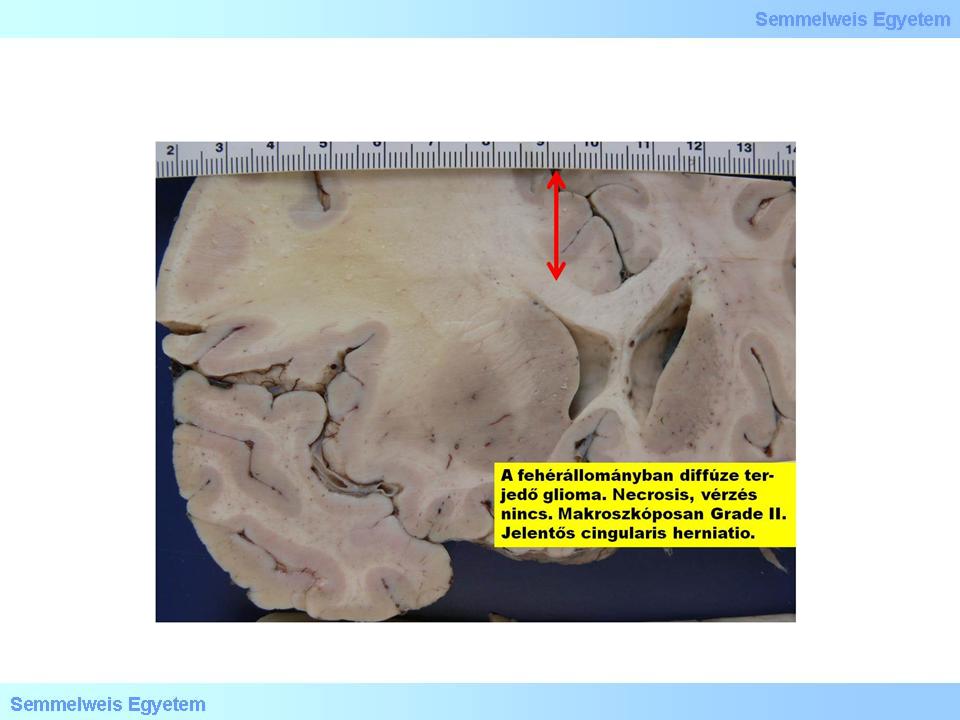
1.Makrofoto: Diffus wachsendes Gliom der weissen Hirnsubstanz. Man sieht keine Nekrosen und Blutungen, weshalb dieser Tumor makroskopisch als Gr II. eingeteilt werden kann - ganz im Gegenteil zum polymorphen Bild der Gr IV. Glioblastome. Es ist eine bedeutende Verschiebung des Gyrus Cinguli und der Mittellinie (s.roter Pfeil) zu sehen (Peter Molnár; Pathologisches Institut des OEC, Universität Debrecen).
|
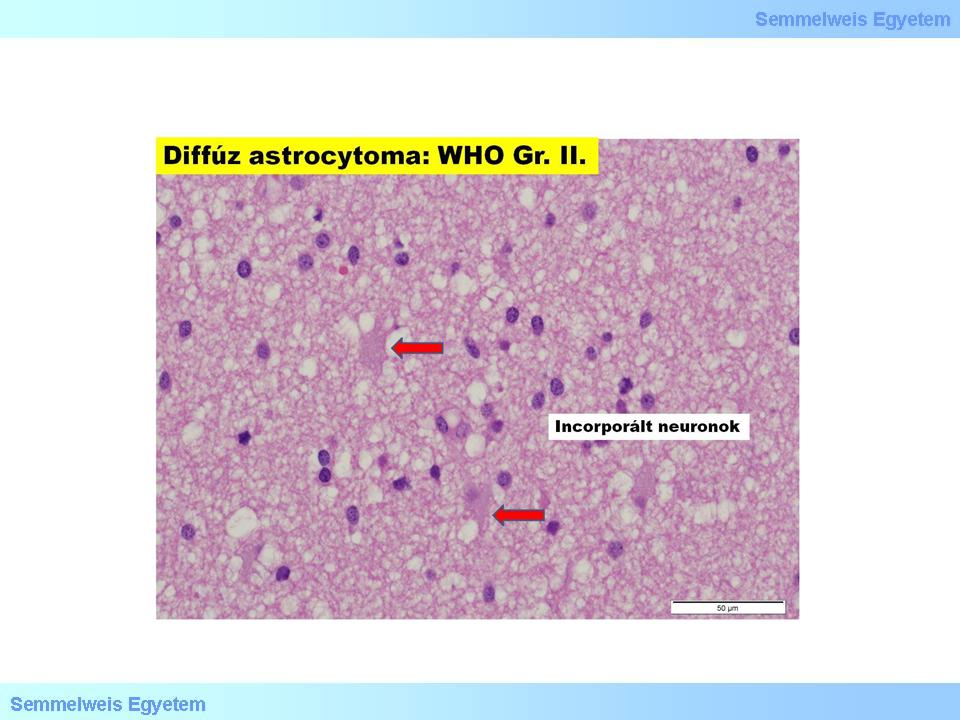
1.Mikrofoto: Diffuses Astrozytom (Gr II.) (Peter Molnár, Pathologisches Institut des OEC, Universität Debrecen).
|
Die biologische Progression dieses Tumors bedeutet, dass er histologisch immer weniger dem Ausgangsgewebe gleicht, und die Malignität, also die biologische Aggressivität des Tumors, zunimmt. So kann es sein, dass ein Gr II. Tumor erst zu einem Gr III. (anaplastisches Astrozytom), und schliesslich zu einem Gr IV. (Glioblastoma multiforme (11A)) Tumor mutiert. Die fibrillären Astrozytome zeigen typischer Weise eine minimal erhöhte Zellzahl (Zellularität), eine leichte Polymorphie der Zellen, ausserdem eine leichte Hyperchromasie der Zellkerne, und die unregelmäßige Anordnung der Zellen im Gewebe, was auf das infiltrative Wachstum dieses Tumors hinweist. Ist bei solch einem Tumor eine leicht erhöhte Zellkernatypie zu sehen, wird er deshalb noch nicht in einen höheren Grad eingestuft. Zellteilungen sind in diesem Tumor kaum, und wenn, dann nur ganz zerstreut zu sehen; Nekrosen und eine Epithelproliferation fehlen.
Das diagnostische Kriterium für ein gemistozytäres Astrozytom ist, dass mindestens 20 % der Zellen Gemistozyten sind. Bei diesen Zellen handelt es sich um aufgedunsene Astrozyten, die nicht neoplastisch verändert sind, sondern reaktive Zellen darstellen, die bei akuten und chronischen Schädigungen des Gehirns vorhanden sein können; sind sie bei einer neoplastischen Läsion vorhanden, wird der Tumor in die Untergruppe der astrozytären Veränderungen eingeleitet (10A). Die Histiozyten haben ein eosinophiles Zytoplasma, in dem randständige Zellkerne (der Name stammt angeblich aus dem deutschen Wort "gemästet") und eine fibrilläre Zellmatrix zu sehen sind. Die Zelldichte (Zellularität) ist etwas erhöht (15P -2A. Mikrofoto). Die Zellen sind bei der immunhistochemischen Reaktion mit GFAP stark positiv (15P -2B. Mikrofoto), und bei der Betrachtung elektromikroskopischer Aufnahmen kann man Massen von intermediären Filamenten und Megamitochondrien sehen. Diese Zellen zeigen einen Hang zur Progression, was zu einem plötzlichen extensiven Wachstum des Tumors führen kann; laut dem morphologischen Bild allein kann man den Zeitpunkt dieses explosionsartigen Wachstums leider nicht genau voraussagen.
|
Beurteilen Sie das Bild!
|
2A-B. Mikrofotos: A: Astrozytom - im mittleren Sichtfeld sind 2 grosse Zellen erkennbar (Gemistozyten). B: Der Tumor ist stark GFAP positiv (Peter Molnár - Pathologisches Institut des OEC, Universität Debrecen).
|
II./2.3.4.4.: Anaplastisches Astrozytom (Gr III)
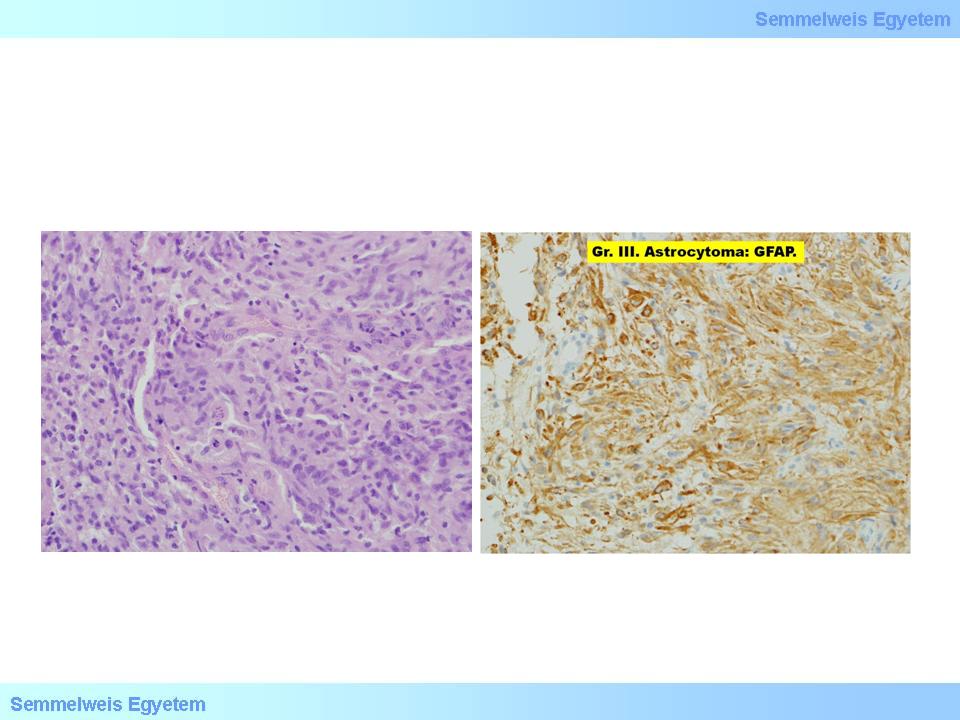
Histologisch wird diese Art von Tumor durch ein diffuses Wachstum mit einer ausgesprochenen Zelldichte, einer Zellatypie, der Hyperchromasie der Zellkerne, und durch eine erhöhte mitotische Aktivität gekennzeichnet. Die bei den anderen Astrozyten auch schon beschriebene Heterogenität ist in diesem Falle noch stärker vorhanden: Unterschiede bei der Form, der Grösse und der Färbung der einzelnen Zellkerne, und eine erhöhte mitotische Aktivität. Die mitotische Aktivität kann heutzutage automatisch mit Phosphohiston3 (PPH3) immunhistochemisch ausgewertet werden (x/1000 Zellen). Die Ki-67 Reaktion, bei der die gegen das Antigen gebildete Antikörper- Menge (Mib-1) bestimmt wird, ist weniger genau. Im Laufe der Zeit wird das histologische Bild immer aggressiver, es kommen mehrkernige Zellen mit bizarren Zellkernen vor, und innerhalb der Zellkerne kann man dann auch Einschlusskörper und prominente Nukleoli, sowie verstreut einige Gemistiozyten sehen.
In dieser Gruppe der Tumore gibt es weder Nekrosen, noch eine Endothelproliferation oder eine Proliferation glomeruloider Kapillaren (Zusammenfassend also keine "mikrovaskuläre Proliferation": MVP). Die Bildung von klonogenen Subpopulationen erkennt man an der Bildung von eng beieinander liegenden / kompakten Zellgruppen, die von ihrer Umgebung nur sehr schwer zu unterscheiden sind. Die Ätiologie einer klonalen Selektion einer Zellpopulation dieses Tumors (also das Überleben von Zellgruppen in einem bestimmten Mikromilieu) ist unbekannt, das Ergebnis dieses Vorganges ist aber, dass diese Zellgruppen die sie umgebenden Tumorzellen, die zwar besser ausgereift sind, aber weniger schnell wachsen, verdrängen. Schliesslich bleibt nur noch die am wenigsten differenzierte Tumorform, (das Glioblastoma multiforme (11A)) übrig, und der komplette Tumor wird dedifferenziert, er unterläuft also einer kompletten onkologischen Progression.
II./2.3.4.5.: Glioblastoma (multiforme) (GB(M), Gr IV.)
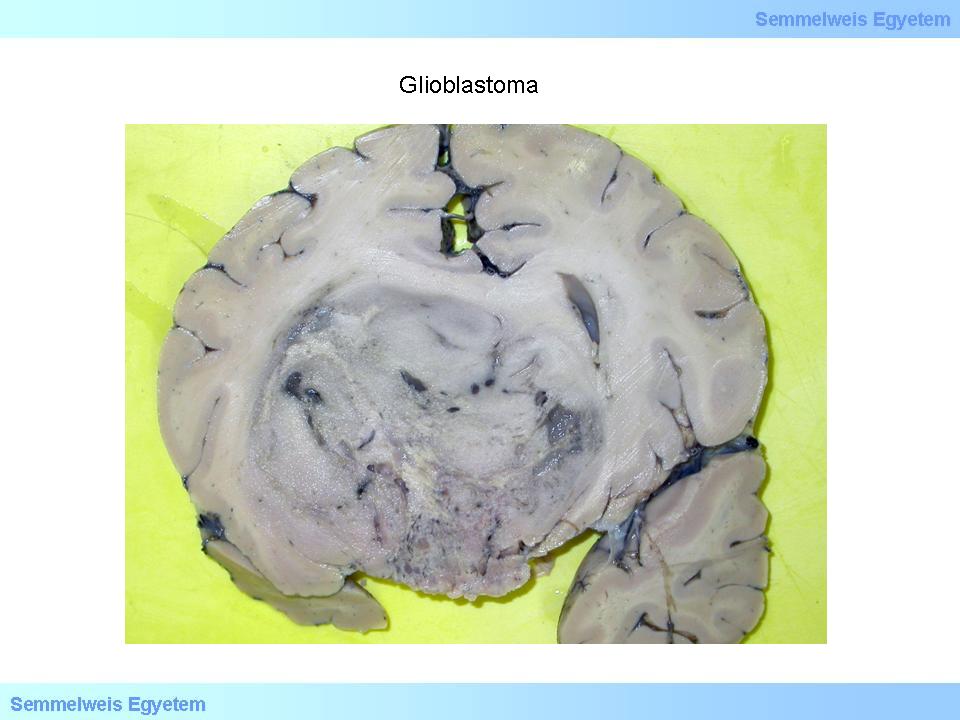
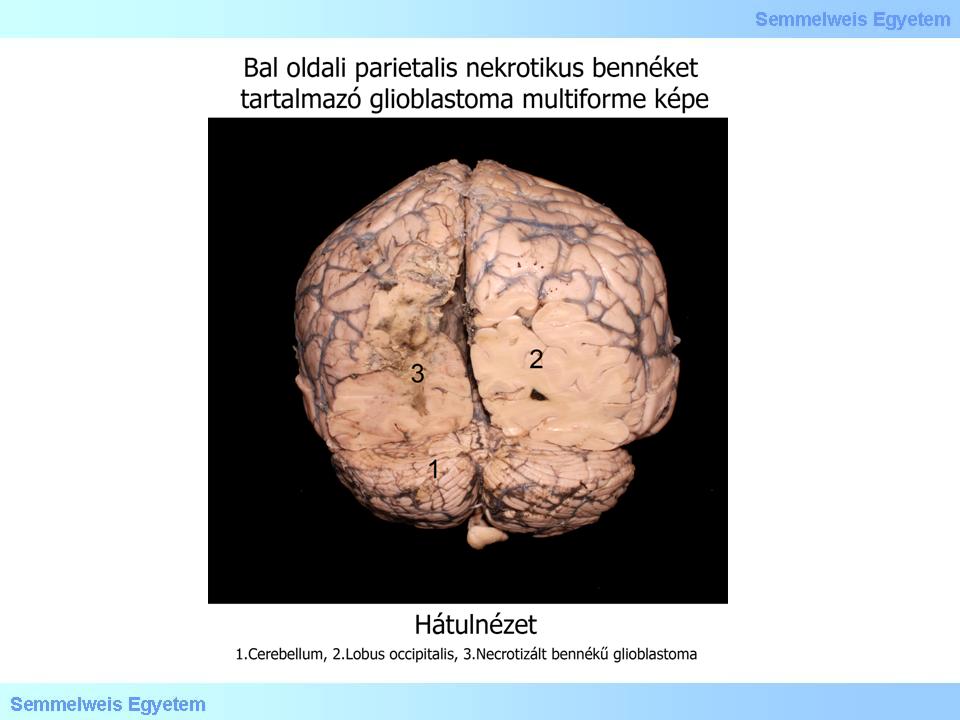
Bei den Glioblastomen handelt sich um die am meisten malignen Tumoren des zentralen Nervensystems, die ohne eine Behandlung innerhalb weniger Monate (6-9) zum Tode eines Patienten führen; aber selbst die modernsten kombinierten Behandlungsmethoden verlängern die Überlebensaussichten eines Patienten kaum etwas, auf maximal 10-14 Monate. Makroskopisch zeigt der Tumor einen gut sichtbaren Rand (2.Makrofoto), obwohl es sich um einen infiltrativ wachsenden Tumor, und nicht um einen metastatischen Tumor handelt. Ausserdem ist das makroskopische Bild, das so ein Tumor zeigt, leicht mit einer Hirnblutung zu verwechseln (3A- C. Makrofoto), da ausgedehnte Nekrosen (2.Foto) das Gefässnetz des Tumorparenchyms zerfressen, und so zu Blutungen führen; klinisch entspricht das Bild dem eines Hirnschlages (Stroke).
|
Beurteilen Sie das Bild!
|
2.Makrofoto: Die makroskopisch sichtbaren scharfen Konturen des Glioblastoma multiforme können zu Verwechslungen führen, da es sich sehrwohl um einen infiltrativ wachsenden Tumor handelt. Die Tumormasse hat das restliche Gehirnparenchym stark deformiert (Peter Molnár, Pathologisches Institut des OEC, Universität Debrecen)
|
2.Foto: Glioblastoma multiforme
|
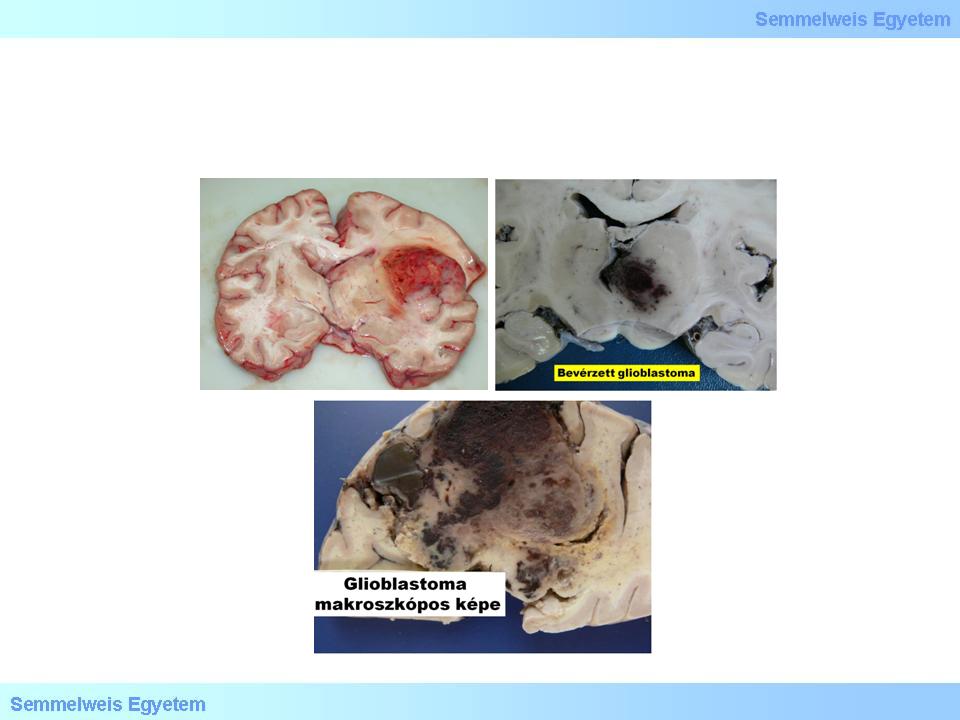
3A-C Makrofoto: Wegen dem Hang einzubluten, zeigt ein Glioblastoma multiforme an der makroskopischen Schnittfläche ein geflecktes Bild, das leicht mit einer Hirnblutung verwechselt werden kann (Peter Molnár, Pathologisches Institut des OEC , Universität Debrecen).
|
Es gibt eine primäre und eine sekundäre Form dieses Tumors. Die erste Form, das Glioblastoma multiforme bildet sich de novo aus; das sekundäre GBM bildet sich durch eine biologische Progression aus einem Astrozytom niedrigeren Grades aus (11A). Ein Glioblastom (der Ausdruck Glioblastoma multiforme wird heutzutage kaum mehr benutzt, weil auch monomorphe Typen dieses Tumors, wie z.B. das kleinzellige Glioblastom (GB), bekannt sind) kommt entweder durch die Proliferation einer pluripotenten Progenitorzelle, oder durch die progressive Dedifferentiation besser ausgereifter Formen zustande. Die von der WHO 2007 festgelegten diagnostischen Kriterien beziehen sich auf den histologischen Aufbau des Tumors: Nekrosen mit den umgebenden Pseudopallisaden (lattenzaunartig angelegte Tumorzellen) und/oder MVP. Ausserdem sind noch die bei den anaplastischen Astrozytomen beschriebenen zytologischen Veränderungen zu sehen. Der vitale Tumor ist sehr zellreich (hyperzellulär), die Zellen sind anaplastisch, undifferenziert (3. Mikrofoto), und befinden sich in einem spongiformen (mikrozystischen) oder fibrillären Grundgewebe.
|
Beurteilen Sie das Bild!
|

3.Mikrofoto: Histologisches Bild eines Glioblastoms: Anisonukleose, Hyperchromasie, Endothelproliferation (Peter Molnár, Pathologisches Institut des OEC, Universität Debrecen).
|
II./2.3.5.: Oligodendrogliome
II./2.3.5.1.: Grad II Oligodendrogliome (OGDG, Gr II)
Es handelt sich hierbei um einen makroskopisch üblicher Weise in einer der Hemisphären lokalisierten Tumor, der oft Verkalkungen zeigt, in der Grosshirnrinde bzw. in der weissen Substanz darunter gebildet wird, und oft auch die Hirnrinde infiltriert. Dies ist histologisch an einer sich um die Nervenzellen ringende Tumorzellinvasion - den sog. Tumor Satelliten - zu beobachten (4. Mikrofoto). Betrachtet man den histologischen Schnitt solch eines Präparates ohne Mikroskop, kann man die Grenze zwischen Rinde und Mark des Gehirns nicht erkennen, und auch die Tumorränder sind sehr verwaschen; die mehrfachen typischen Verkalkungsherde und die häufigen intratumoralen Blutungen sind allerdings gut zu erkennen. Die Schnittfläche des Tumors ist oft geleeartig, glänzend (4. Makrofoto), was auf eine "myxoide Degeneration" zurückgeführt werden kann: einerseits wird dieses Material von Oligodendroglia Zellen gebildet, die eine schleimige, PAS positive Masse akkumulieren (Letztere werden Mukozyten genannt), andererseits wird auch in der oft mikrozystisch veränderten interzellulären Matrix Schleim abgelagert, was dieses makroskopische Aussehen verursacht (13A).
|
Beurteilen Sie das Bild!
|
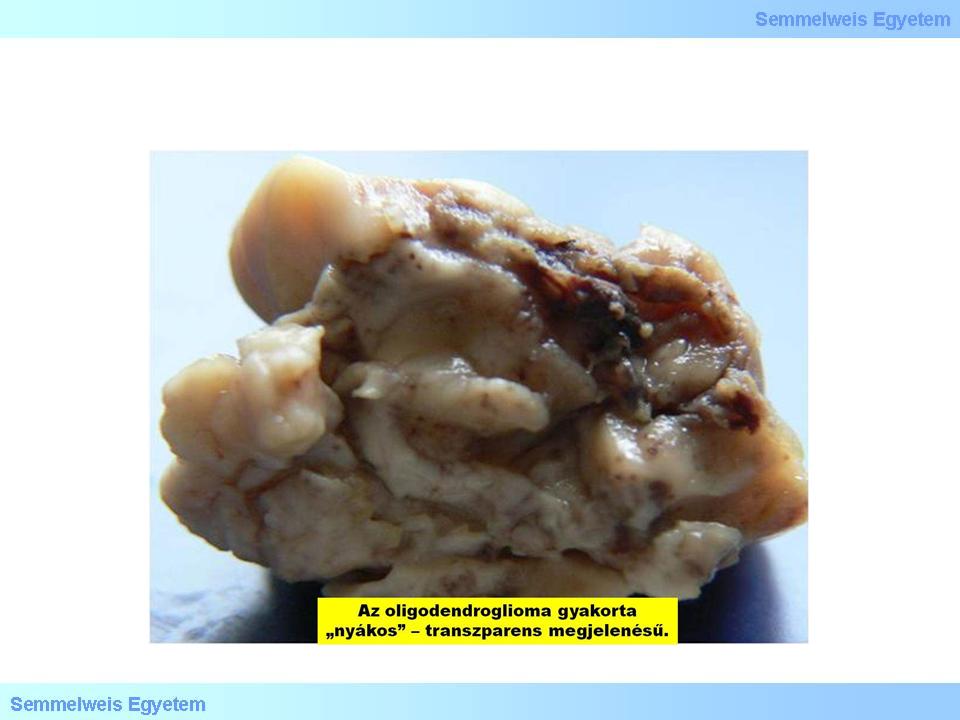
4.Makrofoto: Oligodendrogliome haben oft eine geleeartige äussere Erscheinung (Peter Molnár, Pathologisches Institut des OEC, Universität Debrecen).
|
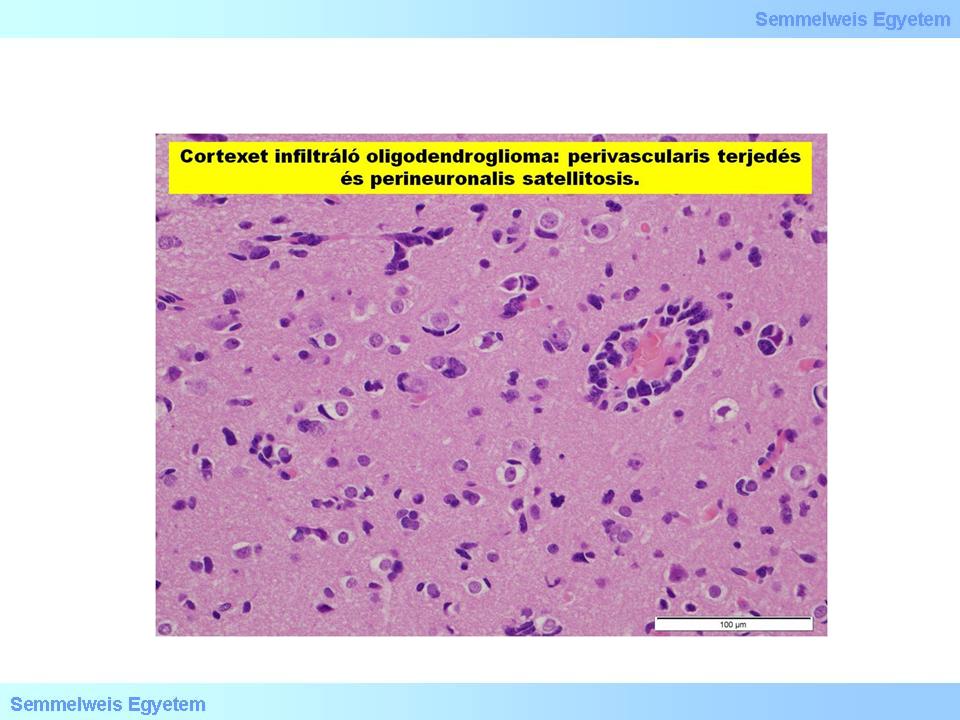
4.Mikrofoto: Oligodendrogliom, das den Cortex infiltriert: man sieht die perivaskuäre Verbreitung und eine perineurale Satellitose (Peter Molnár, Pathologisches Institut des OEC, Universität Debrecen).
|
Das Vorhandensein sog. "Minigemistozyten" ist für die Diagnosestellung ausschlaggebend: man sieht Zellen mit einem hell eosinophilen Zytoplasma und exzentrischen Zellkernen, die im Durchschnitt kleiner sind als die oben beschriebenen Gemistozyten. Auch bei den Oligodendrogliomen ist eine stetige Dedifferenzierung unumgänglich, was heisst, dass die als differenziert geltende Gr II. Tumore zu malignen, aggressiv wachsenden anaplastischen Formen, also zu Gr III. Tumoren werden können; aus diesen Tumoren bildet sich aber nie ein Glioblastoma multiforme (11B). Das histologische Bild der Gr II. Tumore sieht folgender Massen aus: man sieht Maschendraht artie Gefässe, monomorphe Zellkerne, um die Tumorzellen herum ein optisch leeres Zytoplasma (sog. perinukleär Halo) und eine extrazelluläre Verkalkung (Bemerkung: der Begriff "halo" stammt aus dem Griechischen, und bezeichnet die Bahn, entlang der die Zugpferde in Kreis herum laufen mussten - und auf diesen Ausdruck ist auch die Benennung der aus der Kunst bekannten Heiligenscheine zurückzuführen!). 50 % der Oligodendrogliome sind GFAP positiv, was darauf zurückzuführen ist, dass diese Tumore in einer bestimmten Entwicklungsphase zur GFAP Produktion fähig sind.
II./2.3.5.2.: Anaplastische (maligne) Oligodendrogliome (Gr III.)
|
|
Die wichtigsten morphologischen Kriterien für ein bereits als maligne geltendes Oligodendrogliom sind die Bildung von Nekrosen, die Verschiebung der Zellkern- Zytoplasma Relation zugunsten des Zellkerns, und eine starke Zellatypie, was alles mit einer Hyperzellularität zusammen vorkommt. Ein weiteres sicheres Zeichen dafür, dass sich der Tumor in eine aggressive Form verändert hat ist, dass sich die Zahl der Mitosen extrem erhöht, und eine vaskuläre Endothelproliferation (MVP) zu sehen ist. Diese Form des Tumors dringt auch oft in die Liquorräume ein - und komischer Weise tun das ausgerechnet die gut differenzierten, absolut nicht anaplastischen Zellen dieses Klons -, was die Lebensaussichten des betroffenen Patienten schnell zunichte machen kann (hydrocephalus internus, herniatio tonsillarum cerebelli).
II./2.3.5.3.: Oligoastrozytome
Die Oligoastrozytome bilden eine gemischte, nur sehr subjektiv bewertbare Kategorie der Gliome, die eine der am meisten diskutierte Streitfrage der Neuro - Pathologie darstellt. Auch bei diesen Tumoren sind zwei verschiedene Grade bekannt (Gr II., Gr III.). Bei Gr III. Oligoastrozytomen (Gr III.OA) entsprechen alle histologischen und zytologischen Charakteristika denen der Glioblastome, es kommen aber zusätzlich noch diffus oder manchmal auch separiert angeordnete Zellelemente dazu, die in einem perinukleären Halo angeordnet sind. Die Beurteilung dieser Kriterien hängt sehr stark von den Erfahrungen und der subjektiven Beurteilung des diagnostisierenden Pathologen ab. Um dieses Problem zu umgehen, hat man versucht, die Kategorie der "Glioblastome mit oligodendroglialen Komponenten" einzuführen. Diese Frage scheint sich aber deshalb zu erübrigen, weil intensive retro- und prospektive Untersuchungen bewiesen haben, dass das Vorhandensein dieser Zellkomponente die Überlebensaussicht des Patienten nicht wirklich beeinflusst (EORTC,2012).
II./2.3.6.: Ependymoma (GrII)
Bei den Ependymomen handelt es sich um Tumore, die makroskopisch i.A. gut umschrieben sind, selten infiltrieren, eine weiche Konsistenz und eine grau-rötlich bzw. grau- weissliche Färbung zeigen, Einblutungen bzw. zystische und nekrotisch Gebiete haben, langsam wachsen und meist von der Wand der Hirnventrikel ausgehen. Im "klassischen" Fall erscheint dieser Tumor im IV.Ventrikel, oder bildet ein intramedulläres Gebilde im Rückenmark. Der Tumor ist in mehrere histologische Typen aufgeteilt. Histologisch sieht man meist perivaskuläre Pseudorosetten, also solch eine Anordnung der Tumorzellen um Gefässe herum, dass deren Ausläufer bis an die Gefässwand reichen, wodurch sich ein perivaskulärer Hof ohne Zellkerne bildet, was beim betrachten des Objektträgers mit blossem Auge so das Bild eines fibrillären, eosinophil gefleckten Gewebes ergibt. Dieser perivaskuläre, fibrilläre Hof ist stark GFAP positiv. Die Betrachtung dieses Präparates mit blossem Auge führt wegen der Dominanz der Pseudorosetten oft leichter zu einer Diagnose, als wenn man es mikroskopisch mit einer grossen Vergrösserung versucht.
|
|
Ein weiteres typisches Zeichen dieses Tumors, das histologisch zur Diagnosestellung verwendet werden kann, ist das Vorhandensein von ependymalen Rosetten. Hier ordnen sich die länglichen Tumorzellen um ein zentrales Lumen herum an, und bilden dadurch sog. "ependymale Kanäle" (ähnlich des embryologisch angelegten primitiven Wirbelkanales). Letztere kommen seltener, die Pseudorosetten etwas häufiger vor. Es ist auch eine anaplastische Form der Ependymome bekannt (Gr III.), bei der man nicht nur histologisch unreife (anaplastische) Zellen sehen kann, sondern auch eine erhöhte mitotische Aktivität, pseudopallisadenartig aufgestellte Tumorzellen, die um Nekrosen herum zu sehen sind, und eine erhöhte Gefässproliferation beobachten kann. Das sog. zelluläre Ependymom stellt eine eigenständige Gruppe dar, bei der eine erhöhte Zelldichte zum normalen histologischen Bild gehört, ohne dass es sich dabei um einen Tumor höheren Grades handelt. Immunhistologisch zeigen die Ependymome eine positive Reaktion mit GFAP-, Vimentin-, Nestin-, manchmal auch mit S-100- und EMA; beim anaplastischen Typ ist auch die Reaktion mit Zytokeratin positiv. Selten kann es bei diesen Tumoren zur Melanin Bildung kommen- wie auch bei anderen neuroektodermalen Tumoen. Eine eigenständige, spezielle Form der Ependymome bildet das tanzytäre Ependymom (Gr II.), und das myxopapilläre Ependymom, dass von vielen als Gr I. beurteilt wird. Letzteres bildet sich meist an der Cauda equina. Der Tumor wächst langsam, kann aber oft nicht in toto entfernt werden.
II./2.3.7.: Zentrales Neurozytom (Gr II)
Es handelt sich hierbei um einen supratentoriell wachsenden, meist intraventrikulär lokalisierten Tumor, der auf einem CT isodens erscheint, Gadolinum anhäuft und oft verkalkt. Der Tumor verschliesst leicht das Foramen Monroi, was zu einem Hydrozephalus führt. Bei intraoperativ angelegten Abdrücken des Tumors kann man sehen, dass die Zellen gleich sind, denen der Oligodendrogliome ähneln, und keine Zellausläufer haben. Ausser den monomorphen Tumorzellen kann man eosinophile, kernlose, fibrilläre Neuropili, und manchmal die Bildung von Pseudorosetten sehen.
II./2.3.8.: Embryonale Tumore
Alle embryonalen Tumore haben eine gleiche Eigenschaft, und zwar die, dass sie aus dedifferenzierten runden Zellen bestehen. Das heisst, dass diese Tumore zur Gruppe der sog. SBTC - small blue cell tumor, der Gruppe der kleinzelligen Tumore gehören. Die Farbe blau im Namen deutet darauf hin, dass sich dieser Tumor bei der HE Färbung stark basophil zeigt; auch bei der Betrachtung eines histologischen Schnittes mit blossem Auge zeigt solch ein Tumorpräparat einen blauen Tonus, da die meisten Zellen, aus denen der Tumor besteht, hyperchrome, vergrößerte Kerne haben, die den grössten Teil des Zytoplasmas besetzen, von dem nur noch ein dünner Rand übrig bleibt. So kommt es, dass der ganze Tumor aus relativ monomorphen Zellen besteht, was monomorphe Atypie genannt wird. Zu den kleinzelligen Tumoren gehören ausser den embryonalen Tumoren (Neuroblastome, Hepatoblastome, Pankreatoblastome, Nephroblastome, usw.) noch die kleinzelligen Lungentumore, die kleinzelligen B-Zell- Lymphome, und das Ewing-Sarkom.
II./2.3.8.1.: Neuroblastome (GrIV)
Es handelt sich hierbei um einen hochgradig malignen Tumor, der in erster Linie in der Kindheit vorkommt. Innerhalb des Schädels ist dieser Tumor meist supratentoriell gelegen, kann aber auch im Kleinhirn vorkommen. Der Tumor kann auch extrakranial gelegen sein, und kommt dann am häufigsten im peripheren Sympathikus (vor allem in den paravertebralen sympathischen Ganglien, im Nebennierenmark und in den Paraganglien (z.B. im Zuckerland -schen Organ)) vor. Bei einem klassischen Neuroblastom dominieren die Neuroblasten (über 50 %) und Neuropil, und man kann auch häufig Neuroblastrosetten sehen. Es wird eine dedifferenzierte Form (bei der auch eine grosszellige, pleomorphe Variation beschrieben wird), eine niedrig differenzierte und eine differenzierte Form unterschieden. Immunhistochemisch zeigt dieser Tumor eine diffuse, starke NSE -Positivität. Sonstige Marker, die bei diesem Tumor positiv sind: Synaptophysin, NFP, Gangliosid GD2, Chromogranin. Der Proliferationsindex liegt zwischen 10 und 80 %.
II./2.3.8.2.: Primitiv neuroektodermale Tumore (PNET, GrIV)
Die PNET- Konzeption stellt in Bezug auf die Einteilung der primären Hirntumore eine der am meisten diskutierte Streitfrage dar. In der Peripherie kommt diese Art von Tumor öfter vor, er ist aber auch schon supratentoriell beschrieben worden, und hat die unterschiedlichsten Benennungen bekommen: zerebrales Medulloblastom, zerebrales Neuroblastom, supratentorieller kleinzelliger Tumor (SBCT- small blue-cell tumor) usw. Die SBCT sind von Zellen umgeben, die in verschiedenste Richtungen spezialisiert sein können (neuronale, astrozytären, ependymale, muskulöse, melanozytäre Differentiation), was diese Tumoren zu einem Sammelbegriff macht. Histologisch sieht man anaplastische Zellen, unter denen oft wirklich die SBCT dominieren, und keinerlei Zeichen einer Differenzierung zeigen. Die Zellkerne sind hyperchrom, das bisschen an Zytoplasma, das noch zu sehen ist, hat eine verschwommene Grenze, und die interzelluläre Matrix ist fibrillär, und kaum noch erkennbar. Der mitotische Index ist sehr hoch (einem Gr IV. entsprechend). Man sieht auch oft Blutungen bzw. Nekrosen im Tumor, und manchmal bilden sich auch sog. Homer- Wright- Rosetten (eine aus hyperchromen Tumorzellen bestehende Formation, die sich um ein helles, aus Neuropil bestehendes Zentrum blütenartig anordnen). Die primitiven Zellen expressieren muskeltypische Antigene, manchmal auch gliale Antigene an ihrer Oberfläche. Ein supratentorieller PNET ist zytologisch von einem Medulloblastom kaum zu unterscheiden.
II./2.3.8.3.: Medulloblastome (GrIV)
Medulloblastome sind typischerweise im Kleinhirn lokalisierte, maligne, invasiv wachsende, embryonale Tumore, die die wichtigsten Vertreter der PNET- Gruppe darstellen. (Die Benennung ist nicht ganz gelungen, weil es ein "Medulloblastom" per se eigentlich nicht gibt). Es kann vorkommen, dass sich der Tumor an der Basis des IV. Ventrikels bildet, und das Lumen der Kammer vollständig ausfüllt; der Tumor selber ist oft rund, hat eine fein granulierte, unregelmässige Oberfläche (5. Makrofoto), zeigt eine relativ eindeutige Tumorgrenze, ist relativ klein, aber trotzdem anaplastisch, und besteht aus Zellen mit hyperchromen Zellkernen und einem minimalem Zytoplasma; Stroma ist in diesen Zellen auch kaum zu erkennen. Das Vorhandensein von Rosetten ist zur Diagnosestellung nicht unbedingt nötig, da sie bei vielen Tumoren gar nicht vorkommen. Dieser Tumor führt i.A. ohne eine solide, homogen akumulierende, kombinierte Chemotherapie schnell zum Tode des Patienten, nicht zuletzt wegen seinem ausgesprochenen Hang zur Infiltration des Liquors, und zur Abgabe von Fernmetastasen entlang der Neuroaxis. Da die häufigste Lokalisation dieses Tumors die hintere Schädelgrube ist, wächst er in der Nähe des Ventrikels, wodurch es oft zur Ausbildung eines Hydrozephalus kommen kann. Immunhistochemisch zeigt sich bei diesem Tumor ein sehr variables Bild: er ist sowohl Synaptophysin, bei Reaktionen mit Neurofilament-spezifischen Proteinen, Nestin, Vimentin, und an einigen sternförmigen Tumorzellen auch GFAP positiv. Der Proliferationsindex liegt oft über 20 %. Die Aufstellung einer Prognose der Krankheit den histologischen Charakteristika entsprechend ist eigentlich unmöglich; das Vorhandensein einer aneuploiden DNS stellt allerdings ein prognostisch gutes Zeichen dar.
|
Beurteilen Sie das Bild!
|
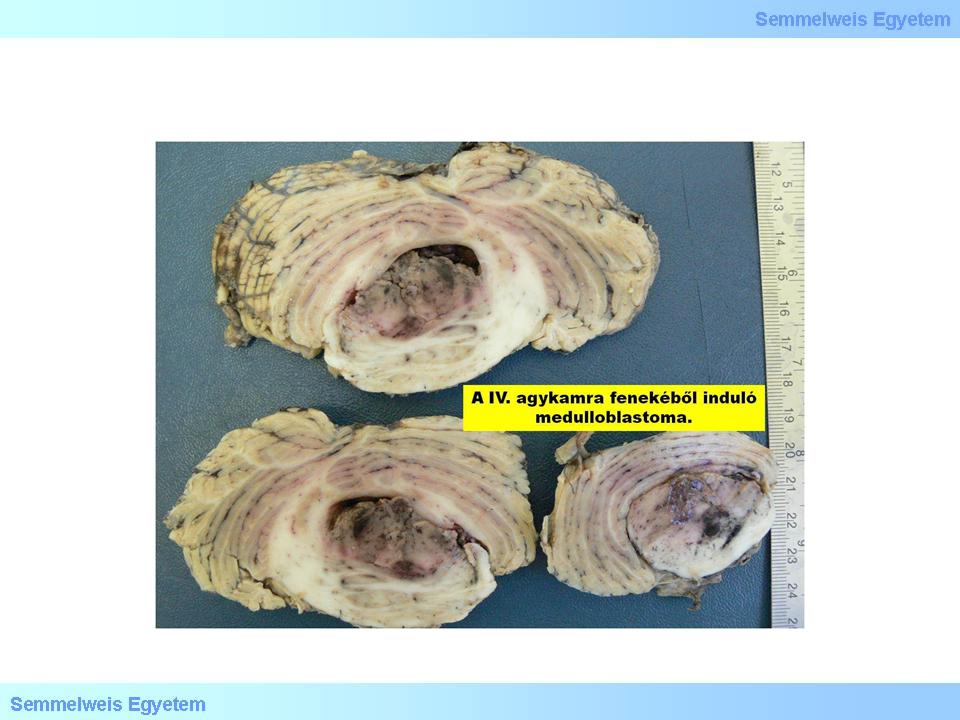
5.Makrofoto: Das Medulloblastom zeigt eine ausgesprochen bunte Schnittfläche (Peter Molnár, Pathologisches Institut des OEC, Universität Debrecen).
|
Die neuesten Forschungsergebnisse zeigen, dass die bisher für eine einheitliche Gruppe gehaltenen PNET- die Medulloblastome und die anderen kleinzelligen Tumoren (SBCT- small blue-cell tumor) (5. Mikrofoto) - genetisch verschiedene Tumore darstellen. Bis zur Jahrtausendwende gab es keine diagnostischen Hilfsmittel zur Unterscheidung dieser Tumore voneinander, und noch weniger solche, mit denen man eine Prognose hätte aufstellen können. Mit der DNS- Microarray Technik ist es allerdings gelungen, das Expressionsprofil der Medulloblastome eindeutig aufzuzeigen (2002), das sich von dem der supratentoriellen PNET und dem anderer anaplastischer maligner Gliome vollkommen unterscheidet. Es ist ebenfalls bewiesen worden, dass die Medulloblastome aus dem Stratum granulare der Rinde des Kleinhirns stammen, und sich dann bilden, wenn das Sonic Hedgehog Regulationssystem (SHH) aktiviert worden ist. Ferner ist festgestellt worden, dass bei der Bildung des desmoplastischen Medulloblastoms die SHH -Rezeptoren (PTCH, GLI) und das N- MYC eine wichtige Rolle spielen.
|
Beurteilen Sie das Bild!
|
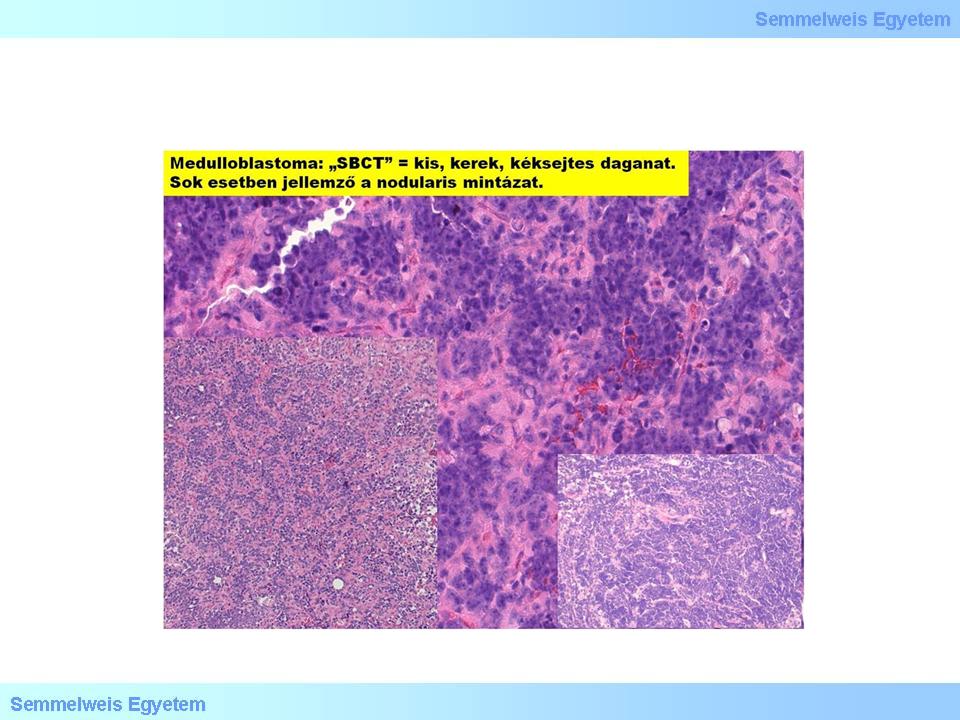
5.Mikrofoto: Das Medulloblastom stellt einen kleinzelligen Tumor dar, der oft ein noduläres Muster zeigt (Peter Molnár, Pathologisches Institut des OEC, Universität Debrecen).
|
|
|