 |
VIII./5. fejezet: Szekvenciaadatok elemzése
Bevezetés
A nyers mérési adatok előállításától kezdődik meg a munkafolyamat következő, szakasza, az adatelemzés. Az újgenerációs szekvenálással hatalmas mennyiségű adat nyerhető. Például az Illumina Novaseq platformjának egyetlen futása akár 6 terabyte-nyi adatot is eredményezhet. Ezt az adatmennyiséget valamilyen módon fel kell dolgozni, majd elemezni és tárolni is kell, ami nagy számítástechnikai teljesítményt és tárhelykapacitást igényel. Számos alkalmazás ún. felhőszervereken működik. Az újgenerációs szekvenálás bioinformatikai munkafolyamata a különböző mérőműszerek esetében detektált jelek adatokká, az adatok értelmezhető információkká és az információ hasznosítható tudássá konvertálását szolgálja. Ennek folyamata három lépésre osztható: elsődleges, másodlagos és harmadlagos elemzés.
|
 |
VIII./5.1. Elsődleges elemzés: a rövid leolvasott szekvenciarészletek előkészítése
A különböző szekvenáló technológiák az eltérő méréstechnikai módszernek megfelelően különböző kiindulási nyers adatokat használnak (képfájlokat vagy éppen feszültség-változásokat), a cél a sorrendezett nukleotidok alkotta kód kimenete. Az átalakítást a szekvenáló készülékek beépített szoftvere végzi. Minden egyes DNS fragmentumra vonatkozó, rövid szakaszokban (short reads) leolvasott szekvenciaadat FASTQ fájlformátumban kerül eltárolásra. A read minden bázisához a szekvenáló platform hozzárendel egy a bázishívás minőségének mértékét leíró, a kapilláris szekvenálás során kidolgozott Phred-like pontszámot. Ez alapján az alacsony minőségű read-eket eltávolítják. Az elsődleges analízis magában foglalhatja a különböző, egy időben futtatott egyének mintáinak megkülönböztetésére a könyvtárkészítés során eltérő oligonukleotid-címkékkel (index vagy barcode) megjelölt fragmensekről készült read-ek szétválogatását (de-multiplexing) is.
|
|
VIII./5.2. Másodlagos elemzés: illesztés a referenciára és az eltérések azonosítása
A másodlagos elemzés kezdeti lépése az illesztés, vagyis a read-ek egy referencia genom komplementer pozícióihoz való illesztése. A read-ek egy folytonos szekvenciává történő de novo összeillesztésére is léteznek megoldások, de ezek nem szükségesek a jól térképezett emberi genom esetében. Miután az illesztés megtörtént, néhány finomító lépés következik, amelyek során például kiszűrik a PCR-reakció alkalmazásából adódó műtermékeket, illetve az elvégzett átrendezés az inzerciók vagy deléciók helyes illesztését teszi lehetővé. Végül a read-ekből összeállított konszenzus szekvenciát egy SAM (sequence alignment/map) vagy egy BAM (binary aligment/map) fáljban tárolják el.

5.ábra Az újgenerációs szekvenálás során leolvasott rövid szakaszok (short reads) a referenciaszekvenciára történő illesztését követő vizualizációja az IGV eszköz segítségével.
|
A következő lépés a variánshívás (variant calling), vagyis az összeállított szekvencia összehasonlítása ismert referencia szekvenciával annak érdekében, hogy meghatározzák, mely pozíciók térnek el a vad típusú allélokat tartalmazó referenciaszekvenciától. Az NGS technológiák viszonylag gyakran ejtenek random hibát, ez azonban rendszerint jól ellensúlyozható a megfelelő szekvenálási mélységgel, azaz egy bázis-pozícióra jutó minél nagyobb számú read-del, ami a pozíció többszöri leolvasását jelenti. Az eltérő pozíciók listája, vagy variánslista egy VCF (variant call format) fájlban kerül rögzítésre.
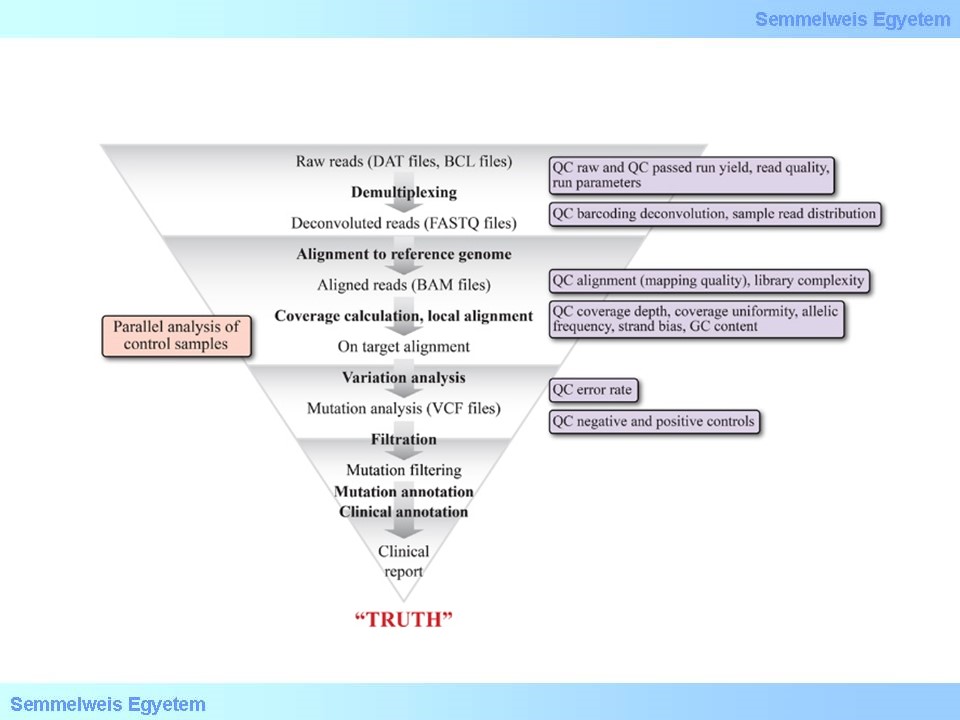
6.ábra A teljes adatelemzési folyamat áttekintése az adatállományok és a minőségi jellemzők számba vételével. (forrás)
|
|
|
VIII./5.3. Harmadlagos elemzés: annotálás és szűrés
A harmadlagos elemzés célja a nagyszámú azonosított szekvencia-variáns közül megtalálni az adott fenotípus (tünetegyüttes) kialakulásáért felelős mutáció(ka)t, illetve a variáns-jelöltek számának lecsökkentése az orvosi döntéshozatalt hatékonyan támogató mennyiségre.
A variánsok felismerését követően azok annotálását kell elvégezni a várható biológiai jelentőségük meghatározása céljából, lehetővé téve ezzel a funkcionális alapon történő prioritizálást és a következő lépésben történő optimálisan súlyozott értelmezést. Az annotálás magába foglalja minden eddig ismert információ lekérdezését az összes észlelt variánsról. Ez a jellemzést különféle biológiai annotációs forrásokból származó gyakorisági, szerkezeti, predikciós és bizonyítékon alapuló adatok kombinációja alkotja.
A fellelt variánsok interpretációja leginkább a nagy mennyiségű adatot generáló vizsgálatok esetében - mint amilyen a teljes exom szekvenálás - jelent kihívást, mivel több tízezer variáns kerül hívásra. Ezek több lépésből álló szűrésétől várható a valószínűleg nem kóroki, gyakori és a megbízhatatlan, rossz minőségel hívott vagy illesztett variánsok kizárása.
Első lépésben az aspecifikus, azaz a célrégión kívül található (off-target) variánshívásokat szűrik ki. Ezt követi a szekvenciaminőség alapján való szűrés, amely segítségével a rossz minőséggel hívott vagy rosszul, azaz kevés read-del lefedett variánshívásokat zárják ki. Harmadik lépésben a populációs gyakoriság szerinti szűrést végzik el, kizárva a gyakori SNP-ket, amelyek összefüggése valószínűtlen az vizsgált fenotípus ritka előfordulási gyakorsága mellett. A kritérium ennél a lépésnél változó, de általánosan azokat a variáns-hívásokat szűrik ki, amelyek minor allél gyakorisága (minor allele frequency, MAF) meghaladja az 1%-ot valamelyik, az emberi populációk haplotípusait kvantitatívan reprezentáló adatbázisban, mint amilyen az 1000 Genomes Project.
Ezen a ponton feltétlenül szükséges a variáns-szintről, ami a genomi pozícióra vonatkozó információ (pl. chr10:g.230845794A>G) a kódoló szekvenciában, a gén-szintre történő átváltás, ami az aminosav-sorrendben elfoglalt helyzetet jelenti (pl. p.Met268Thr az AGT génben). Ez teszi ugyanis lehetővé a variáns típusának, várható következményének felmérését a géntermékekre gyakorolt hatás alapján. Itt vizsgálható meg, hogy a feltételezett kórképek esetében leírták-e már ezt a pozíciót, illetve ismert-e a klinikai jelentősége a betegségadatbázisok alapján.
|
|