
A teljes exom szekvenálás az esetek 40-50%-ban hozza meg a genetikai diagnózist.
|
XV./3. fejezet: Teljes exom/genom szekvenálás
Az emberi exom az összes kódoló nukleáris DNS-szekvenciát magába foglalja, amely körülbelül 180.000 exont, összességében mintegy 30 megabázisnyi érett RNS-re is lefordítódó szekvencia adatot tartalmaz. A mitochondriális DNS-ben található kódoló szakaszokat nem soroljuk az exom fogalmához. Jóllehet az exom az emberi genomnak csak 1% -2% -át teszi ki, az exonokban található meg a jelenleg ismert betegség-okozó variáns többsége. Utóbbihoz persze hozzátartozik, hogy az aminosav-cserére, szerkezetre, funkcióra gyakorolt hatásokat is könnyebb értelmezni. Az exom szekvenálás célja a genomban lévő összes fehérjét kódoló nukleáris gén szekvenciájának leolvasása és elemzése. Az összes exoni szekvencia körülbelül 95% -a határozható meg a jelenleg rendelkezésre álló technikákkal. A klinikai célú exom szekvenálás diagnosztikai haszna mintegy 30-40%-ra tehető, az öröklődőnek imponáló, de a genetikai kórok feltárására irányuló célzott vizsgálatokkal sikertelenül diagnosztizált egyének szisztematikus vizsgálata alapján.
Az elmúlt néhány évben az exom szekvenálás egyre inkább elérhetővé vált, hiszen egy teljes exom szekvenálás laboratóriumi önköltségét akár 300 USD is fedezheti. Ez a technológia a klinikai gyakorlatban a mendeli öröklésmenetet mutató kórképek hátterében álló pontszerű genomi eltérések (pontmutációk és kisebb inzerciók-deléciók) feltárásában használható fel, utóbbi időben a nagyobb kópiaszám elemzések is megvalósíthatóak. A klinikusnak ismernie kell azokat a genetikai rendellenesség típusokat, amiket az újgenerációs szekvenálás nem ismer fel.
Önmagában a labor technológia azonban nem elegendő ahhoz, hogy klinikailag fel tudjuk használni a keletkező óriás mennyiségű információt. Fejlett bioinformatikai módszerek és pipeline-ok kellenek a teljes exom és genom adatok klinikai értelmezéséhez.
Az adatelemzési lépésben nyert annotált variánslista exom szekvenálás esetében mintegy 20,000 variánst tartalmaz, azonban panel szekvenálás esetén is, a lista több százas nagyságrendű lehet. A ritka betegségek vizsgálatakor általában egy-két kauzális mutáció azonosítása a célunk, így a variánsok számának szűkítésére további bioinformatikai és manuális lépésekre van szükség (2. Ábra).
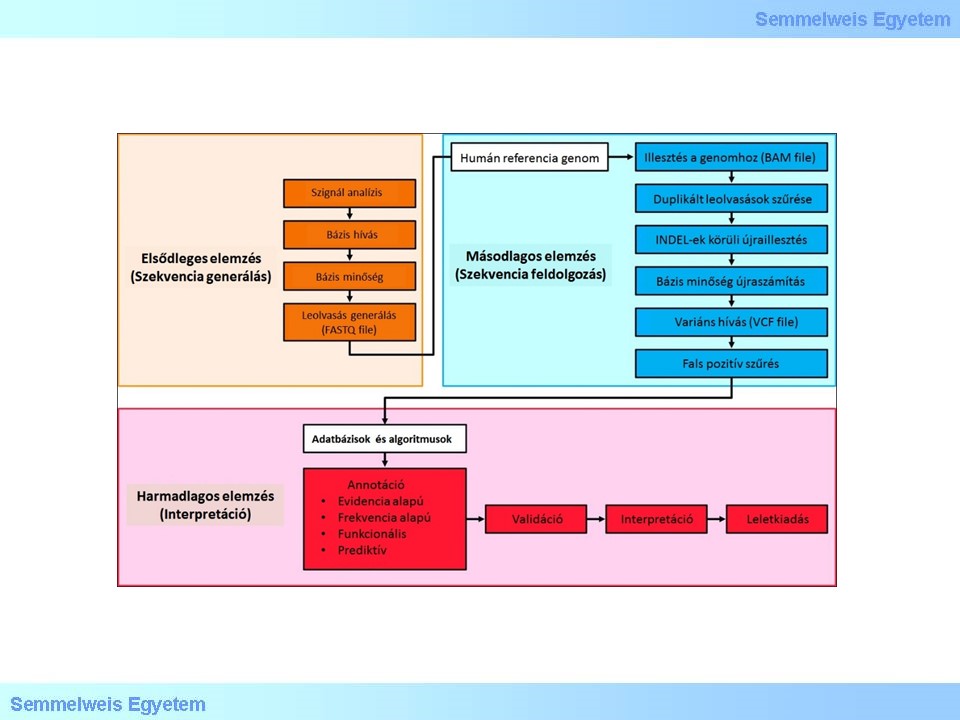
2. ábra: A klinikai NGS analízis fő komponenseinek folyamatábrája
|
Nehézséget okoz, hogy a variánsok jelentős hányada ismeretlen jelentőségű. Még a gyakran vizsgált BRCA1/2 gén esetében is az irodalomban közölt variánsok ~20%-a ún. bizonytalan szignifikanciájú variáns („variant of uncertain significance”, VUS), a kevésbé gyakran vizsgált génekben ez elérheti az50%-ot is.
A variánslista szűkítésére tehát különböző szűrési feltételeket alkalmazunk, amelyek általában a következő elveken alapulnak:
-
1) Öröklésmenet alapján történő szűrés.
-
2) Evolúciós konzerváltságot mérő, valamint a biokémiai struktúra változását prediktáló szoftverek pontszámai.
-
3) Pathogén variánsok listáját tartalmazó adatbázisokban történő keresés.
-
4) Populációs illetve belső adatbázisokból származó minor allél frekvencia adatok.
-
5) Fenotípus alapú szűrés.
|
 |
A predikciós szoftverek gépi tanulási eljárásokat alkalmaznak annak becslésére, hogy a nukleotid, vagy aminosav változás mekkora valószínűséggel okoz a fehérjében károsodást. Mivel a különböző predikciós szoftverek eltérő jellemzőkkel bírnak, és eredményeik csak részben átfedőek, ezért általában ajánlott több predikciós szoftver alkalmazása. A minor allél frekvencia adatok szintén fontosak a ritka betegségekben végzett genetikai vizsgálatok esetében, azonban figyelembe kell venni, hogy az egyes adatbázisokban nem csak egészséges egyének adatai jelenhetnek meg. Az elmúlt években az adatelemzésben szerepet kapnak a fenotípust is figyelembe vevő, variáns szűrést támogató szoftverek. Ehhez elengedhetetlen a fenotípus standardizált, számítógép által feldolgozható leírása, amelyre a humán fenotípus ontológia (HPO) ad többek között lehetőséget. A végső lépésben, minden esetben szükség van a talált variánsok kutató, illetve genetikus orvos általi értékelésére. Az NGS kísérletek elterjedésének köszönhetően egyre több variáns került közlésre az irodalomban, így szükség volt a variánsok kategorizálásának egységesítésére. Az American College of Medical Genetics and Genomics (ACMG) ajánlása alapján a variánsokat, megszabott kritériumok szerint, 5 osztályba soroljuk (patogén, valószínűleg patogén, benignus, valószínűleg benignus, ismeretlen szignifikanciájú). Napjainkban a ritka variánsok legtöbbje a bizonytalan jelentőségű csoportba tartozik.
|
 |
A mai rutin diagnosztikában akkor alkalmazzuk ezt a vizsgálatot, ha egy klinikailag nagyon homogén tünet együttes hátterében rendkívül sokféle gén, génhálózat tagjainak érintettsége is feltételezhető, pl. mentális retardáció, macrocephalia, stb. olyan esetben, ahol igen nagy a valószínűsége, hogy genetikai betegségről van szó. Esetenként nagyon szerteágazó tünetcsoport esetén is megkísérelhetjük ezt a diagnosztikai eljárást, ha genetikus betegségre gondolunk. Intenzív osztályon kezelő csecsemőknél, ha a klinikai kép és a laborok alapján nincs jó céldiagnózisunk és feltételezzük az örökletes betegséget, sokkal költséghatékonyabb és gyorsabban célra vezet egy WES vagy WGS vizsgálat számos tanulmány szerint, mint az egymásra épülő szekvenciális vizsgálatok és ráadásul nem ritka, hogy terápiás következménye is van a megszülető diagnózisnak. Azokban az esetekben, ahol jól azonosítható a klinikai kép, pl. spastikus paraparesisek, örökletes neuropathiák, agyi vastárolással járó kórképek a tergetált, vagy panel szekvenálást részesítjük előnyben (1. ábra)
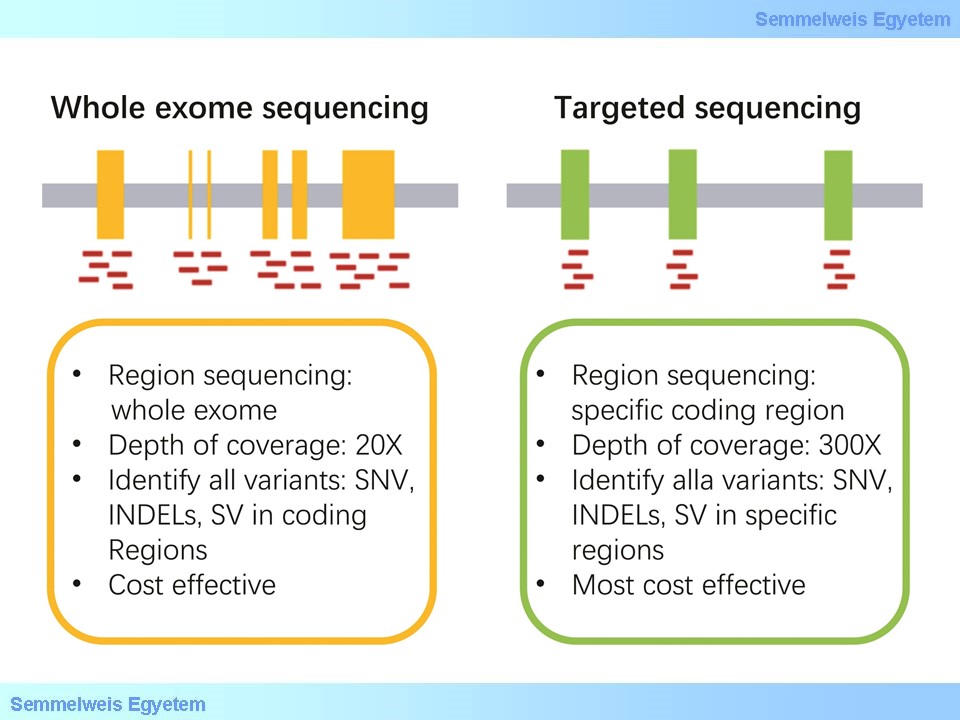
1. ábra: A Panel (targetált szekvenálás) és a teljes exom szekvenálás összehasonlítása
|
|
WGS ma még csak kutatási projektekben érhető el. A nem exoni regiók értékelése jelenti a legnagyobb nehézséget, ugyanis az introni szakaszokról sokkal kevesebb információval rendelkezünk, mint az exonikaról.
|
Mik a nehézségei és a veszélyei a WES és WGS vizsgálatnak?
Egy WES vizsgálat során több tízezer ritka variánst fogunk azonosítani és a missense variánsok száma is meghaladja az ezret. Különböző komputeres predikciós programmal értékeljük ezeket a variánsokat. A programok figyelik, hogy mennyire konzervált a hely, ahol a variáns keletkezett, milyen mértékben károsíthatja a fehérje funkcióját a mutáció. Ezen kívül azt is figyeljük, hogy milyen gyakran fordult elő eddig a variáns az egyes nagy populációs adatbázisokban. Minél kisebb a minor allél frekvencia, annál nagyobb a valószínűsége, hogy betegséggel asszociációt mutató variánsról van szó. Egy – egy ilyen variáns azonosítását követően az irodalmi adatokban rá kel keresni, hogy az adott mutációt emberi betegséggel társulva leírták-e, és ha igen, akkor a fenotypus egyezett-e? Figyelni kell azt is az elemzés során, hogy az általunk vizsgálat betegben autoszomális recessiv vagy domináns öröklődésmenetről van –e szó. Az esetek alig 1-2% ban találunk biztosan patogén mutációkat. Hasonló az aránya a valószínűleg patogén mutációknak. A variánsok csaknem 50%-a a bizonytalan jelentőségű variáns kategóriába esik. A biztosan benignus és valószínűleg benignus mutációk száma is relative alacsony. Az egyes mutációk besorolásához az American College of Medical Genetics and Genomics (ACMG) ajánlását használja a világ nagy része.
Azokban az esetekben ahol bizonytalan jelentőségű variánsokat találunk csak, nem tudunk a páciensnek és családjának biztosat mondani a betegség etiológiájáról. Ilyen esetben szegregációs vizsgálatok illetve kutatási projektek keretén belül funkcionális vizsgálatokkal lehet igazolni az adott variáns patogenitását. Ez a mindennapi gyakorlatban a talált összes VUS vonatkozásában nem valósítható meg. A mai ismereteink szerint a WES vizsgálatok kb. 30-40% os valószínűséggel igazolja a betegség hátterét, a WGS vizsgálatok találati aránya kissé magasabb.
|
|