| |
I./2.4.: Other developmental anomalies of the kidney
I./2.4.1.: Renal agenesis
|
 |
Absolute lack of renal development during embryogenesis. In case of bilateral renal agenesis, oligohydramnios appears due to the lack of renal functions. Before birth, the embryonic blood gets detoxicated through maternal blood but soon after birth, uremia appears and the newborn dies, if the anomaly has not been detected earlier. In those cases, when fetal ultrasound examination reveals this anomaly, the pregnancy has to be ceased. In case of unilateral renal agenesis, renal dysfuntion do not occur since the contralateral kidney takes over and compensates for the missing functions, with hypertrophy (hypertrophia compensatorica seu vicarians).
I./2.4.2.: Renal hypoplasia
The size of the kidney is significantly smaller. Usually it affects only one kidney, while the other kidney shows compensatory hypertrophy. It can be differentiated from the secunder atrophy of a well-developed kidney (atrophia renis), based on the number of pyramids and papillas, which is <8 in hypoplasia. This condition increases the vulnerability to infections too, and chronic pyelonephritis leads to hypertonia.
I./2.4.3.: Oligohydramnios
Fetal urine gets into the amnial space, and the majority of the amniotic fluid, that is necessary for the undisturbed development and free movement of the fetus, is from fetal urine. If there is not enough amniotic fluid, the necessary space for the fetus is not provided, and the tension of the muscular wall of the uterus directly affects the fetus, causing compressional deformities: compressed face and skull, parrot-like face, undeveloped mandible, hypoplasia of the lungs in the compressed chest, hip dislocation, deformities of the limbs may occur.
I./2.4.4.: Congenital hydronephrosis
The kidney is enlarged, the parenchyma is atrophic, calyces and the pelvis are large, filled with urine, becuase the pyeloureteral junction (junctio pyeloureteralis seu ureteropelvicalis) is stenotic (stenosis pyeloureteralis). It is mostly unilateral, and if recognized early by ultrasound, the kidney can be saved by cutting out the stenotic part.
I./2.4.5.: Kidney dystopia
Usually this condition affects the left kidney, which is positioned lower, than the level of Th11-L3 vertebrae, occasionally it is located in the pelvic cavity. The renal artery (a. renalis) branches from the lower third of the aorta, or from the iliac artery (a. iliaca). The ureter is short, broken, or compressed, or it can possibly join pathologically to the bladder. Forms: dystopia abdominalis (kidney is positioned at the level of the L2-3 vertebrae); dystopia abdominalis pelvica (kidney is positioned at the pelvic inlet – promontorium); dystopia pelvica (kidney is in the pelvis). Its complications are associated with the disturbed leak: hydroureter; hydronephros; pyelonephritis]; nephrolithiasis; atrophia renis.
I./2.4.6.: Duplicated pelvis and ureters
|
 |
Urine is drained by two pelvises and two ureters from one kidney. In this condition, the kidney is often divided into two, at its middle third, by a transversal parenchymal septum. One of the two ureters usually joins the bladder pathologically, and thus, the upper parts of the urinary tract enlarges.
I./2.4.7.: Angiogenetic anomaly
There are more than one renal artery supplying the kidney. The aberrant branches (aa. aberrantes) reach the renal parenchyma directly, separated from the hilus. In unfavorable cases, they can compress the ureter, causing hydronephros.
|

Have a look at the illustration!
|
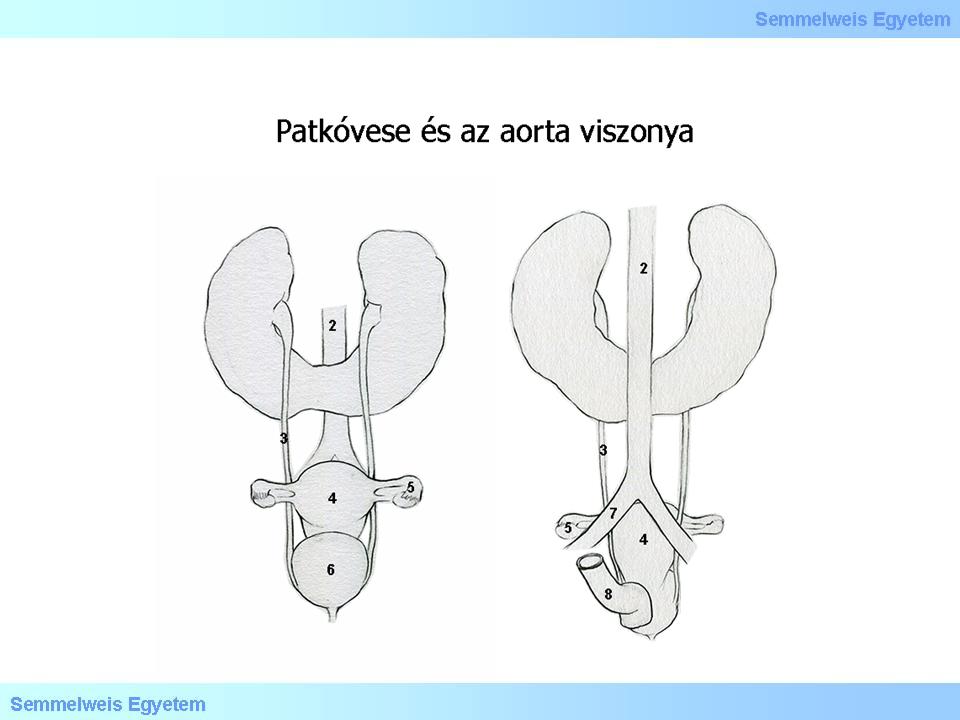
Illustration 3: Patkóvese és az aorta viszonyát magyarázó ábra: (1) Fusio renorum, (2) Aorta, (3) Ureter, (4) Uterus, (5) Ovarium, (6) Vesica urinaria, (7) Arteria iliaca communis, (8) Rectum, (9) Elölnézet
|
I./2.4.8.: Developmental, multicystic dysplastic kidney
It is caused by the unorganized renal development, due to the dysfunctional maturing of the metanephros. It is the most frequent cystic kidney disease in children, and it is the most frequent space occupation in the abdominal cavity, in newborns. Mostly it is unilateral, but occasionally it can be bilateral either, and both focal and segmental forms may occur. It can co-occur with different lesions, at different levels of the urinary tract, such as uretero-pelvic, ureteral, and urethral obstruction, or atresia. Simultaneously, dysfunctions in other organs, especially in the heart can occur.
The severity of the clinical picture is determined by the degree of the dysplasia and the obstruction of the urinary tract. It is typically a sporadic condition, however, rarely, it can be familial either, in which cases it co-occurs with multi- organ dysfunctions. Macroscopically, renal parenchyma is occupied by cysts in a variety of size and quantity; histologically, cysts are lined with cuboidal epithelium, the stroma in between is constituted of undifferentiated elements, often with dysplastic mesenchymal components, with chondroid and fibromuscular islands.
I./2.4.9.: Polycystic kidney disease
|
 |
Two forms are known based on the type of heritability:
-
1) autosomal dominant (adult type), and
-
2) autosomal recessive (infantile type) polycystic kidney disease.
I./2.4.9.1.: Autosomal dominant (adult type) polycystic kidney disease
In this autosomal dominantly heritable condition, with a practically 100% penetrance rate, renal cysts invade the renal parenchyma in a gradually increasing manner, eventually leading to renal failure, in the fourth-fifth decade usually. This is one of the most frequent heritable diseases, it occurs 1-2‰ of live births, and 10% of conditions requiring dyalisis.
|
|
Both genders are equally affected, symptoms include pain in the sides, or even palpable tumescence, haematuria, hypertonia, renal failure, occasionally kidney stones. Although, the disease is bilateral, the occurrence can even be shifted in time, between the two sides. In developed cases the kidneys are enlarged, lost their reniform shape, their surface is tuberculous due to the cysts. In progressed cases, original renal tissue can only be found microscopically. Cysts can appear as diverticulums of any parts of the glomerular capsule and the renal tubules, and over time, they separate from their base. After this, the originally glomerular filtrate may only szaporodhat tovább, as transepithelial secretum.
Histologically, cysts are lined with flat, cuboidal epithelium, the remainder of the intact renal parenchyma in between the cysts is prone for scarring, chronic inflammation, tubular atrophy. This disease can be taken as systemic, since in more than half of the patients, cysts appear in other organs as well, for example in the liver, pancreas, spleen, corpus pineale, seminal vesicle, lung. Interestingly, similar lesions may develop in other sites too: brain- and coronaria aneurysma, mitral valve prolapse, diverticulosis of the colon, skeletal malformations. The genetic lesion transmitted by the parents is almost always one of two mutations: PKD1 (locus: 16p13.3; frequency: 85-90%) and PKD2 (locus: 4q13-23). Approximately, in 10% of the patients, the family history is negative, these cases obviously carry sporadic, new mutations.
I./2.4.9.2.: Autosomal recessive (infantile type) polycystic kidney disease
Contrary to the autosomal dominant form, this type is much less frequent (0.05‰ of live births), and the cystic enlargement of the kidney, as well as the abdominal space occupation, is obvious already at the birth. A further difference is that next to the kidneys, the liver is always affected either, while in the dominant type it is not. The lesions of the liver can be cysts, congenital fibrosis, biliar dysfunctions, and ductectasia, all of which early lead to portal hypertension, hepatosplenomegaly, esophagal varices. All this is accompanied by the specific Potter-phenotype, due to the oligohydramnios, that is facial deformation, joint dysfuntions, pulmonary hypoplasia,.
|
|
Those showing pronounced lesions, die shortly after they were born, usually due to respiratory failure. In those, who survive the perinatal phase, renal failure, systemic and portal hypertension develop. Mortality rate at the perinatal period is 30-50%, after this it is only 5-20%. Macroscopic features also differ from those in the dominant type. Although kidneys are enlarged, their renal (bean) shape is maintained. On the cut-surface, cysts originating from the dilatations of the collecting tubules, by fluid retention, are elongated and fan-like, reaching toward the cortex from the medulla. They are lined with flat cubical epithelium. According to the severity of the condition, intact nephrons may be found among the cysts.
I./2.4.10.: Nephroblastomatosis, nephrogen remains
|
|
These lesions are mostly congenital dysontogenic in nature, rather than neoplastic, however, they are in some kind of histogenetic relation with Wilms-tumor, since they can be found in 30% of Wilms-tumor cases, and in these cases, the mutation of WT1 can be found in both lesions. They appear in one or both kidneys, as one or more primitive metanephric tissue nodule, or they can appear also by forming massive tissue-fields, in which blastema, tubules and stroma are present in different ratio, as well as packed lines of non anaplastic nephrogenic epithelial cells.
|
|