| |
I/2.3.: Arteriosclerosis (hardening of arteries, arterial calcification), as the most important disease predisposing to myocardial infarction
I/2.3.1.: Definitions
|
 |
Arteriosclerosis (an umbrella term for disorders leading to hardening of arteries) is a degenerative alteration of arterial walls. It typically presents with plaques. Plaques are degenerative intravascular wall thickenings that typically occur in arteries. New components, mainly connective tissue fibers, fatty and/or calcified materials appear at a demarcated part of the vascular wall, and – depending on the stage of severity – they:
-
- modify the structure of the vascular wall,
-
- alter its mechanical properties and – over a certain volume –
-
- constrict the vascular lumen.
-
1. Vascular wall modificaton means that the plaque forming material or tissue excess first deforms the intima by thickening it, then it extends to the media, and by eliminating these layers, it modifies the original vascular wall structure.
-
2. Mechanical property alteration means that the arterial wall loses its original elasticity at the plaque site: in some cases – fibrous and/or calcified plaques – it becomes harder and more rigid, while in other cases – large atheromatous plaques – the wall gets softer, and may bulge later, therefore the vessel can less and less act as a wind-box and resist the pulsating load exerted by the blood flow.
-
3. Plaque growing and luminal narrowing means that excess materials and tissues occur as actual space occupations, and as they grow towards the vascular lumen, they gradulally restrict the vessel’s transmissivity.
Within arteriosclerosis, there is a distiction between atherosclerosis which is the disease of elastic and medium size muscular arteries, and arteriolosclerosis which affects small muscular arteries and arteriolas. Mönckeberg’s medial sclerosis is a special and rare form of arterial hardening. These vascular alerations occur especially frequently and complexly in some metabolic diseases, and primarily in diabetes mellitus. Special complications include local bulging of the arterial wall (so-called aneurysms) and the separation of the wall layers (artery dissection).
|
 |
I/2.3.2: Epidemiological significance
Arteriosclerosis occurs as a cause of death in 50% of the cases in the more developed countries (so-called civilization disease) . As opposed to the classic conception which said that arteriosclerosis was the disease of the elderly, these vascular alterations start to develop in young age. First reports of this were published in the 1950s in the United States, based on autopsy reports of young soldiers who were killed in the Chorean war.
I/2.3.3: Lipid metabolism
Lipid metabolism plays a basic role in the development of arteriosclerosis. The characteristics of the most significant lipids in the body’ lipid homeostasis are listed in Table 1.
|

Study the table and analyze what you see!
|
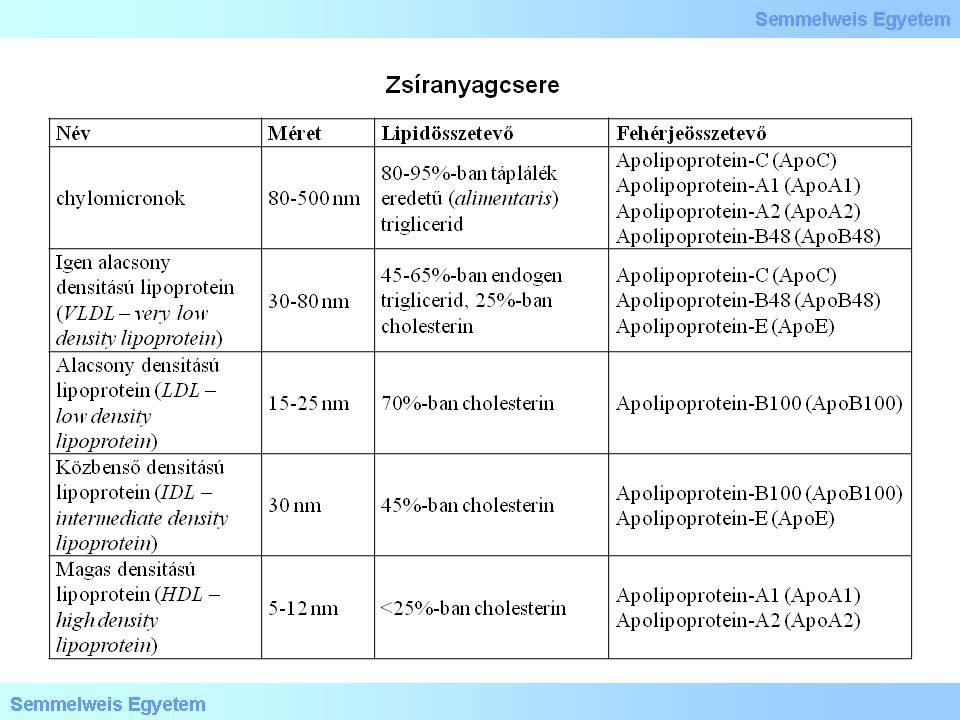
Table 1.: Lipid metabolism
|
The so-called exogenic pathway of these lipids is heading from the bowels to the liver, and is basically a centripetal route; the lipids carried this way are chilomicrons. The centrifugal part of the lipid transport’s so-called endogenic pathway carries the lipids in the form of VLDLs from the liver to the vascular system, and then to the peripheral (extrahepatic) tissues, while lipoprotein lipase turns them into IDLs and LDLs. During centripetal transport, lipids are carried from the peripheral tissues to the liver as HDLs, IDLs and LDLs.
I/2.3.4: Cellular elements of the sclerotic vascular lesions
|
|
The most important cellular elements of arterial hardening are endothelial cells, monocytes/macrophages and smooth muscle cells.
I/2.3.4.1: Endothelial cells
The most important function of intact endothel is to provide a non-adherent and non-thrombotic inner surface. It also regulates the vascular wall’s permeability, the microenvironmental circumstances for fibrinolysis and coagulation, leukocyte and platelet adhesion (i.e. homing), and lipid oxidation. It also produces so-called chemoattractants and extracellular matrix components, exerts proliferating and migrating effects on smooth muscle cells, and has metabolic activity (e.g. LDL oxidation), maintains vascular elasticity and tonicity through its vasoactive products (NO, PGI2, endothelin, angiotensin II, TXA2), maintains vascular wall structure by e.g. producing basement membrane, and participates in inflammatory and immunological reactions.
I/2.3.4.2: Monocytes/macrophages
|
 |
These cells are of central importance in atherogenesis. They participate in lipid metabolism by oxidation, by incorporating extracellular fat, and by producing foam-cells. They produce remarkably large amount of cytokins, like growth factors (GM-CSF, M-CSF, HB-EGF, IGF-I, VEGF, bFGF, TGFα, PDGF, IL-2), growth inhibitors (IL-1, TNFα, TGFß), and chemoattractants (MCP-1, oxLDL).
In the defence system, their substantial role is to present antigens to T-cells and to eliminate and neutralize harmful substances by their ‘scavenger’ effect.
I/2.3.4.3: Smooth muscle cells
Smooth muscle cells have two different phenotypes:
-
a) contractile and
-
b) synthetic.
The contractile phenotype is the generally known cell variation which is rich in myofilaments and is able to contract. Its lipid uptake and depletion is balanced, lipid accumulation doesn’t occur, and this type doesn’t turn into foam cell.
Cells of the synthetic phenotype, on the other hand, are rich in rough endoplasmic reticulum (RER), produce extracellular matrix components upon cytokin stimuli (PDGF, bFGF, TGFß), and express LDL- and ‘scavenger’ receptors which capacitate this cell type to uptake more fat then what’s depleted, to consequently accumulate lipids, and at the end, to transform into foam cells. Smooth muscle cells are able to migrate from the tunica media to the intima.
I/2.3.5: Atherogenic theories
|
 |
There are mostly only theories concerning the development of arterial hardening. The main reason of this is that the pathogenesis is really difficult to model experimentally, since this disease is multifactorial, and it takes long years through decades to develop. The most comprehensive explanation of our latest conceptions is the ‘response to injury’ theory, insomuch as the other theories give only partial answers by emphasizing some elements of this main theory, and the suggestions of these partial theories fit well into the ‘response to injury’ theory. ‘Lipid theory’ accentuates the vascular wall’s lipid metabolism disorders. ‘Thrombogenic theory’ is one of the oldest theories, while ‘inflammatory or infective, autoimmune theory’ emphasizes inflammatory recactions of various origins.
I/2.3.5.1: ‘Response to injury’ theory
|
|
The essence of the theory is that atherogenesis is a stereotypical response of the vascular wall to any kind of harmful impact. Its central process is focal endothel activation (stimulation), and resetting its reactivity and metabolism as the first step of atherogenesis and plaque formation.
I/2.3.5.1.1: Vascular wall damaging effects, their nature and background
Harmful impacts can be of various types, like – among others – cellular biological, geometrical, microanatomical (so called ‘progression-prone’ vs. ‘progression-resistant’ sites), mechanical-hemodynamical, blood chemical, or microenvironmental; and sometimes these impact types occur in combination.
|
|
Hem-mediated cytotoxicity is a recently discovered ubiquitous endothel damaging mechanism belonging to the blood chemical and cell biological type. It causes one type of endothel damage, namely local oxidative stress. Hemoglobin leaking from red blood cells oxidates in the plasma, and ferri- or methemoglobin is formed while hem escapes from its protein coat. The free hem or hem-Fe is toxic to the endothel – because both of them are able to catalyse free radical reactions and thus potentiate cell damage through, for instance, activated polymorphonuclear granulocytes (PMN) (aggravation of oxidant related cytotoxicity by cell mediated cytotoxicity). Endothelial cells in the range of the escaped hem are especially endangered.
The damage is done by the hem which is very hydrophobic, and gets through every cell membranes unobstructedly, and therefore it penetrates endothelial cells easily and spontaneously. The relased hem-Fe is a very active catalyst of cell membrane component and transient LDL molecule oxidation. The nascent oxLDL provokes the endothelial cells’ direct cytotoxicity and oxidative stress. Defensive proteins prevent the above processes to get loose in a self-reinforcing way. These proteins iclude
|
|
-
a) hemopexin, which is a serum protein in extremely high concentration and great hem-binding capacity. The following proteins have the same effect:
-
b) haptoglobin and albumin, as also plasma proteins with hem-binding capacity. A special intracellular protein-cooperation is seen with
-
c) hem-oxygenase-1 (HO-1) and ferritin: HO-1 breaks the hem’s protoporphyrin ring, and release its active iron content, and the antioxidant iron-storing ferritin binds it.
An important determinant of the processes is the systemic-constitutional background where they take place. This includes first of all the host’s genetic constitution and the chemical composition of blood components. The latter includes mainly blood properties like pH value or atherogenic fat saturation. The host’s genetic constitution’s importance is based on the observation that the dynamics of each complicated pathological processes that develop as a web of steps and correlations – like atherosclerosis – is influenced by the functional effectivity of the individual steps’ components (here: factors, transport molecules, enzymes, etc.). Change in the effectivities might even alter the whole process by accelerating or slowing the process part performed by the given component.
|
|
Since the effectivity of a process part can be influenced by several factors – for the participating proteins, for instance, aminoacid sequence determined by the genetic code and configuration which is influenced by the microenvironment, etc. – and the whole pathological process is constituted by several process parts, therefore the disease develops as the result of almost infinite number of impacts on the host level, and thus it opens the door to fine-tuning of development through the function of the individual molecules.
It is therefore possible that several people develop quite severe arteriosclerosis in a quite young age even without the presence of well defined syndromes (with a trivial expression, these patients are‘prone to vascular diseases’), while others have youthful vascular status even in old age; and that atherogenic predisposition manifests in a certain environment – social, nutritional, life style, etc. –, while it remains silent in another (vid. change of epidemiologic data of immigrants in a foreign country). A well functioning host organization system is able to maintain its functional balance even amidst harmful factors, while in less stable biological status, the functional balanced gets damaged even in a mild risk situation, and its maintenance needs special attention (in the first case the individual ‘can eat whatever he/she wants’ while in the other case they need, for instance, low-cholesterol diet).
I/2.3.5.1.2: Hemodynamical factors of plaque formation
At vascular bifurcations (so-called flow dividers) the original laminar flow breaks, and turbulence emerges at the section right after the separation of the blood flow. Although with decreased shear forces, but mainly by the rotating-circulating movement of blood components, their flow speed is decreased above these arterial wall sections, and thus the atherogenic blood components get longer impact time. It is a simple pathological observation that atherosclerosis is typically more severe at vascular bifurcations.
I/2.3.5.1.3: The essence of the therory
|
|
Endothel stimulation provoked by the harmful impact leads to modification in these cells’ lipid metabolism (oxLDL production!) and production of biologically active molecules (chemoattractants, mitogens, growth-regulators, NO, adhesion molecules, thrombogenic factors). As a response to all this, circulating cellular elements – leukocytes, platelets, monocytes, T-lymphocytes – adhere to the activated endothelial cells’ surface (homing), and they leak into the intimal space through the intercellular gaps, accumulate there, and produce migration, mitogenic and growth factors themselves, therefore attracting more and more biologically active cells to the site.
A further consequence of endothelial stimulation is that the permeability of this layer increases, thus the leakage of lipids and cellular elements into the intima increases (insudation). The intima’s cellular richness is further increased by the immigration of medial smooth muscle cells from the other side, through the internal elastic membrane, and as a response to the microenvironment’s citokin effects, the originally intimal smooth muscle cells start to proliferate too. The increased smooth muscle cell content is determinative for plaque formation because their transition from contractile to synthetic phenotype leads to extracellular matrix component (mainly collagen) production.
In the same time, the accumulated monocytes turn into macrophages, and together with the synthetic smooth muscle cells, they start to incorporate extracellular lipids, therefore both cell types turn into foam cells in large amounts. A part of foam cells die of this unlimited lipid uptake, and their intracellular fat content converge into extracellular fat ponds, and their citokins getting into the environment after their death attract more cellular elements. The process becomes self-reinforcing like an avalanche, and this leads to plaque formation.
I/2.3.5.2: Lipid theory
|
|
It considers lipid acccumulation (insudation) into vascular wall layers as the central pathogenetic phenomenon. The importance of this is supported by several epidemiological and clinico-pathological observations, such as:
-
a) correlation between body weight and arteriosclerosis;
-
b) the risk of plaque formation is higher in case of high seLDL and seVLDL, and low seHDL levels;
-
c) the effect of atherogenic diet in animal experiments;
-
d) the effect of hereditary fat metabolism disorders on the vascular system (low number or lack of receptors, gene damages, familial hyperlipidemia).
The essence of the theory is that lipid materials leak into the intima where simultaneously appearing monocytes/macrophages start to incorporate fat, but their special LDL receptors get saturated in time, and aspecific, so-called ‘scavenger’ receptors latch on to lipid uptake, allowing almost unlimited fat incorporation. Since the rate of uptake thus exceeds the cell’s metabolic capacity, the excess is stored in the form of fat drops, and the cell turns into a foam cell. The product of pathological fat metabolism is oxLDL which contributes to endothelial stimulation via oxidative stress. The endothel, however, produces adhesion molecules, and attracts even more monocytes to the site.
I/2.3.5.3: Thrombogenic theory
|
|
It considers vascular wall thrombosis formation (not necessarily occlusive) as a central pathogenetic phenomenon, which deteriorates by further circulating cellular element accumulation, and then the thrombus gets organized thus forming the developed plaque. According to our present kknowledge, thrombosis is rather a consequence than the first step of atherogenesis.
I/2.3.5.4: Inflammatory-infectious-autoimmune theory
In a not irrelevant part of the cases the arterial hardening can’t be explained by conventional risk factors. Therefore there is intense research for non-conventional or alternative risk factors, which raised the attention on the inflammatory infiltration found in greater or lesser amount in the sick parts of the arterial walls. In the pathological background of these infiltrations, mainly latent infectious origin was suspected, and sterile inflammation was also suggested as an immune response or autoimmune process. These stipulations were further supported by the general histopathological observation, that there is almost always some accompanying lymphocyte presence around arteriosclerotic lesions.
|
|
These researches were successful if we take into consideration that some infectious agents can be proved indeed to be present with varying frequency. However, no satisfactory answer was found to the significant question whether the pathogens really are active causative agents or aggravators of the process, or only ‘innocent bystanders’ (so-called innocent bystander hypothesis). Anyhow, anti infectuous (antibiotic) therapeutic series that were introduced to eradicate pathogens and thus heal sclerotic arterial lesions, were not obviously successful maybe due to the multifactorial nature of atherogenesis.
The most research of all pathogens aimed Chlamydia pneumoniae (year of discovery: 1989), which is a highly specific Gram negative bacteria, an obligate intracellular parasite since it doesn’t have an energy-producing system on its own. Its intracellular location hides it from the immune system and – similarly to the best known member of the Chlamydia genus, C. trachomatis (year of discovery: 1907) – it can cause a typically latent, chronically long lasting inflammation with no acute shubs and inducing connective tissue proliferation and scarring (vid. fibrous plaque). Besides the vascular system, the pathogen is typically found in the airways.
Further pathogens that were examined are Helicobacter pylori (it was never seriously supposed to have a causative role in atherosclerosis), CMV, herpes simplex virus, Mycoplasma pneumoniae, and hepatitis-A virus. The common characteristic of these pathogens is that all of them are intracellular infectious agents. They make the originally antiinflammatory, anticoagulant and antiatherogenic microenvironment of the vessels turn into proinflammatory (by provoking humoral and cellular reactions), procoagulant and proatherogenic.
The researches suggested autoimmunity to have a role too. The discovery of so-called anti-cholesterol antibodies (ACHA) and detection of higher serum levels of it in arteriosclerotic patients as compared to the normal population might suggest that the atheromatous core of sclerotic plaques containing cholesterol might become a target for an autoimmune condition during a lasting vascular disease, and that it might deteriorate its progression.
I/2.3.6: Risk factors
Based on the above information, risk factors of arterial hardening can be divided to
|
|
-
- conventional and
-
- ‘new’, non-conventional (alternative) groups.
The most important conventional risk factors are the following:
-
- hyperlipidemia, hypercholesterinemia
-
- diabetes mellitus
-
- smoking
-
- obesity
-
- hypertension
-
- male sex, old age
-
- hyperhomocysteinemia
-
- hereditary, genetic factors.
The ‘new’, non-conventional (alternative) risk factors take effect by the vascular wall’s inflammatory response:
-
- vascular infection caused by pathogens
-
- immune response, autoimmunity.
I/2.3.7: Morphology of arterial hardening
I/2.3.7.1: Anatomical sites of incidence
Anatomical sites of incidence of severe arteriosclerosis in a decreasing order of frequency
|
|
-
1) infrarenal part of abdominal aorta/iliac arteries,
-
2) proximal parts of coronary arteries,
-
3) thoracic aorta/femoral artery/popliteal artery,
-
4) internal carotid artery,
-
5) intracranial vessels: vertebral artery; basilary artery; middle cerebral artery.
I/2.3.7.2: Appearance types of arterial hardening (AHA classification)
AHA (American Heart Association) classification distinguishes early and late arteriosclerotic lesions, and defines only one transitional plaque type between the two groups, the preatheroma. Early vascular lesions typically develop in the first two-three decades of life, they are reversible vascular wall lesions , and they don’t cause any clinical symptoms . However, late stage lesions develop by later decades of life; they are symptomatic, and cause irreversible vascular wall damage .
I/2.3.7.2.1: Early arteriosclerotic lesion – AHA I
It is characterized by mostly submicroscopic changes; these phenomenons are sometimes invisible by light microscopy. Intracellular lipid accumulation (lipid droplets) occurs in some sporadic cells, but these cells don’t congeal into cell groups or individual cell mass yet. It can develop even in early childhood in the so-called progression-prone vascular parts (e.g. vascular bifurcations), where late stage lesions, complicated plaques grow in later life stages.
I/2.3.7.2.2: Early arteriosclerotic lesion – AHA II
It is still characterized by intracellular lipid accumulation, but the cells congeal into a mass (histiocyte masses). This form can be seen macroscopically, this is the so-called fatty streak (macroscopic image No.1).
|
Study the image!
|
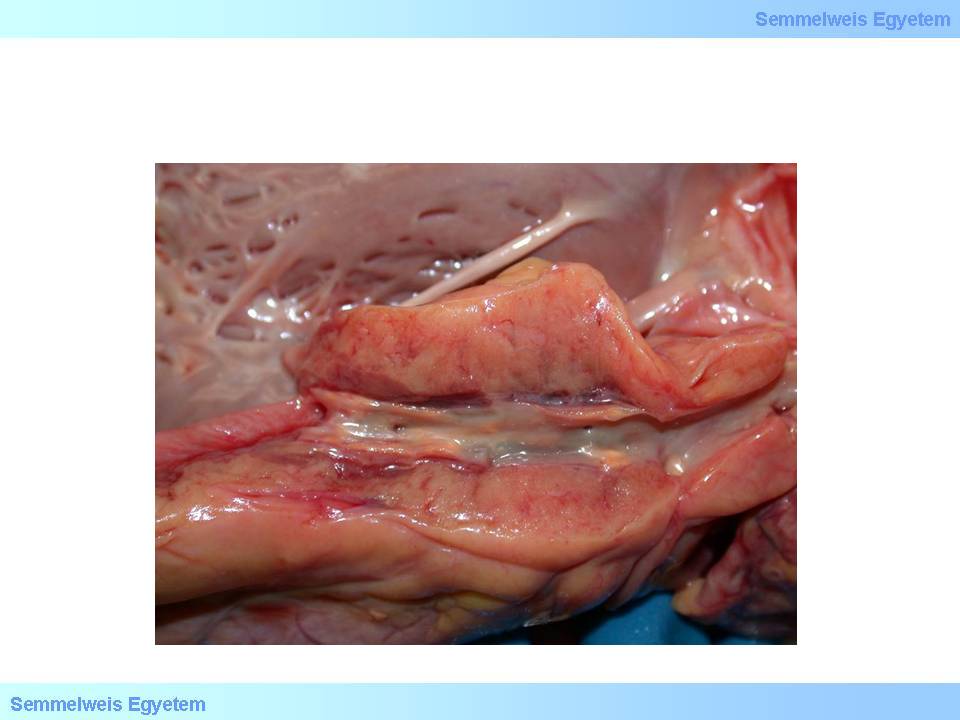
Macroscopic image No.1: Fatty streak as an early and mild arteriosclerotic lesion in a carotid artery. This plaque form doesn’t cause significant stenosis. (From the archives of the 2nd Institute for Pathology, Semmelweis University – collected by Tibor Glasz)
|
I/2.3.7.2.3: Intermediate arteriosclerotic lesion – AHA III
This transient group includes only one member: the so-called preatheroma. It is characterized by both intra- and extrecellular lipid presence: so-called lipid-pools appear. It is also a transition between early and late vascular lesions both morphologically (concurrent presence of intra- and extracellular lipid accumulation) and chemically (in its fatty acid composition).
I/2.3.7.2.4: Late arteriosclerotic lesion– AHA IV
This is the so-called atheroma (athera /gr./ – mush), the unstable development pathway of late stage plaque. Its components are: atheromatous lipid core, often with precipitated, scythe-stone shaped cholesterol crystals, and the fibrous cap covering the lipid core towards the vascular lumen, which is covered by the vessel’s endothel lining. The plaque’s stability is determined by the ratio of components (see there). Calcium salts may deposit in the fibrous cap’s connective tissue. If it extends very deep, the atheromatous lipid core can affect the whole cross-section of the media, thus it significanly weakens the vascular wall’s mechanical properties, and the wall might gradually bulge due to the permanent inner pressure of the blood flow, and form a bag or spindle shaped dilation, the so-called aneurysm.
I/2.3.7.2.5: Late arteriosclerotic lesion – AHA V
Its other name is fibroatheroma (fibrous plaque), it is the stable development pathway of late stage plaque. As compared to atheroma, it consists of a significantly smaller (minimal) atheromatous lipid core and a thick fibrous cap giving the vast majority of the plaque. The lipid core may as well be totally missing, in this case the plaque consists of merely scarred connective tissue. If calcium salts deposit in the plaque (macroscopical image No.2.), it indicates bradytropic circumstances.
|
Study the image!
|
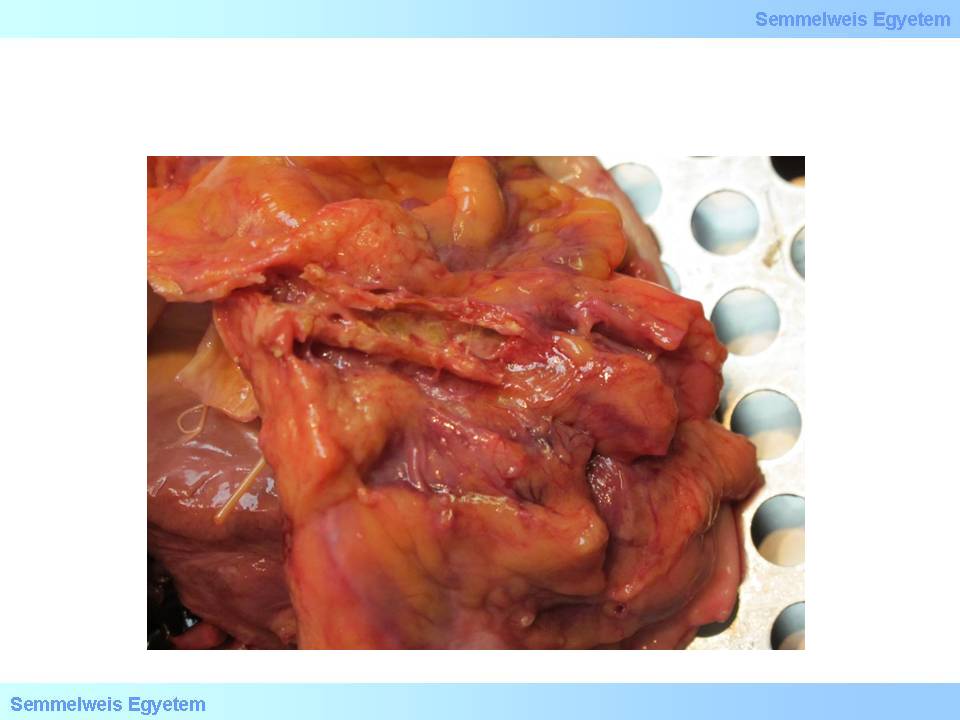
Macroscopic image No.2: Calcified plaques in a coronary artery. Calcification might occur in the tissues in each stages of plaque development as a sign of degeneration. (From the archives of the 2nd Institute for Pathology, Semmelweis University – collected by Attila Kovács and István Kenessey)
|
I/2.3.7.2.6: Late arteriosclerotic lesion – AHA VI
This is the so-called complicated plaque, which develops typically from unstable plaques. There are two main forms of complication: intraplaque hemorrhage and plaque thrombosis.
|
|
-
1. In case of intraplaque hemorrhage the fibrous cap breaks at a demarcated place and opens the way for blood flow into the plaque, into the plastic atheromatous lipid core. The blood streaming into the lipid core (jet phenomenon) inflates the plaque like a balloon, and closes the lumen. A similar plaque rupture and blood infusion can also lead to seaparation of the arterial wall layers (dissection).
-
2. In case of plaque-thrombosis the blood flow brushes off the fibrous cap in total (so-called usurated plaque), and the very thrombogenic lipid core gets free and in direct contact with the blood, and by initiating the extrinsic coagulation pathway it leads to blood clot formation within a short time.
I/2.3.7.2.7: Plaque stability
|
|
By plaque stability we mean the predictability of its morphological-clinical-biological behaviour. Stable plaque is an arteriosclerotic wall thickening with no sudden events or changes expected in its mechanical, structural and concurrent clinical-biological behaviour, therefore its status is stable and predictable. Its typical representatives are the fibrous plaque and fibroatheroma. Unstable plaque is a wall thickening with barely predictable mechanical, structural and concurrent clinical behaviour, and sudden events (so-called plaque complications) and/or morphological changes can’t be excluded [9B], therefore its status is unstable, and unpredictable. Its typical representative is an atheroma with a large lipid core.
By the plaque’s clinical-biological behaviour we mean its effect on the affected vessel’s blood flow: if the plaque is able to cause unexpected fluctuation, and mainly decrease, then the plaque is called unstable, while if the plaque’s presence doesn’t threaten with sudden obstruction of the blood flow, then the plaque is called stable. The plaques’ clinical-biological, and hemodynamic behaviour is strongly related to their morphological composition and mechanical resistance which directly derives from that.
|
|
In clinical terms, the unstable plaque’s hazard lays in its mechanical plasticity, that – depending on the actual hemodynamic situation – is able to change its form suddenly, this morphological variability leads to reciprocal change in the lumen, and this can cause such an unfavourable situation that within a short time, the remaining lumen narrows in a critical degree, that is in a degree that exceeds the ischemic tolerance of the tissues, and it leads to ischemic symptoms (e.g. pain – see angina pectoris, abdominal angina, intermittent claudication, etc.) in the supplied area.
The plaque’s structural stability is therefore in strong relation and direct proportion with its clinical stability, which means that if a plaque is stable structurally and mechanically, then it will also be stable in its clinical behaviour, and vice versa. So, direct conclusions can be made from the plaque’s structural composition to the prospects of the supplied tissues and thus to those of the patient.
In the stability point of view, plaque development can be summarized shortly as follows:
Its earliest significant form is:
-
(1) the fatty streak, which turns into the indermediate form:
-
(2) praeatheroma, and through this it can develop towards two directions:
-
(3-a) unstable plaque (atheroma), and through this stage into
-
(4) complicated lesions (intraplaque hemorrhage and plaque-thrombosis); or towards
-
(3-b) morphological variations of the stable plaque developmental pathway (fibrous-calcified plaque, fibroatheroma)
Risk factors of plaque instability are:
-
1) the plaque’s structure;
-
2) inflammatory infiltration in the fibrous cap;
-
3) calcification of the fibrous cap.
1. In accordance with the above information, the excess of mechanically non-resistant, ‘soft’ components (primarily the atheromatous lipid core) increases the plaque’s plsticity and unstability, and vice versa: the higher the proportion of mechanically resistant tissue components – primarily connective and scar tissue –, the more stable the plaque will be.
2. The presence of inflammatory cells damages the underlying structure, and weakens the fibrous cap forming the ‘lid’ of the plaque via their lytic enzymes, therefore promoting plaque damage.
3. If calcium salts deposit in the fibrous cap, the calcification forms a knife-like plane, and its sharp edge can simply cut into the tissue of the fibrous cap during the plaque’s and the vascular wall’s fluctuating movements, thus weakening it mechanically, and therefore it also contributes to plaque damage.
|
Study the content of the reference!
|
Among other factors increasing plaque instability, first of all we need to consider the fact that accelerated blood flow in the narrowed vascular section leads to locally decreased intraluminal pressure (Bernoulli’s principle) (KV_I_2_fejezet_2_hivatkozas), and this exerts a kind of suction force on the ‘back’ of the plaque, the fibrous cap. Inversely, in the pre- and poststenotic sections, the flow is slower and the inner pressure is higher, therefore the peristenotic wall sections move outwards. The mobility difference between rigid wall sections and the neighbouring, elastic vascular wall parts (see the vessels’ wind box function) also adds to this. All these effects lead to increased traction at the edge of the plaque, and increase the risk of plaque injury, rupture, hemorrhage or even the separation of the wall layers (dissection).
I/2.3.7.2.8: The organs’ reserves against arteriosclerosis
When sclerotic vascular stenosis develops slowly, it provides time to the body to develop mechanisms that help the affected tissues to survive, that is to avoid the area’s losing in total (infarction) – it usually happens at the expense of such compromises, like interstitial fibrosis which forms in chronic ischemia. This is the ischemic tolerance of the affected tissues in case of slowly developing hypoxia, which is supported by the enlargement of the connections (anastomoses) between veessels, and the formation of otherwise insignificant collateral vascular connections.
|
|
Glagov’s compensatory enlargement of arteries means that concurrently with plaque formation, the affected arterial sections enlarge, their outer circumference grows, and this can compensate the plaque’s narrowing effect until the plaque’s size reaches 40% of the lumen, that is, no substantial stenosis occurs until this proportion. This data correlates well with the fact that until the plaque size reaches 50% of the lumen (that is, 50% stenosis), the stenosis is called mild. In case of extreme starvation or weight loss, the plaques which are rich in fat, shrink.
I/2.3.7.2.9: Clinical forms of arteriosclerosis
|
|
The most important clinical symptom of arterial hardening is that the affected organ’s or tissue’s circulatory disorder is followed by functional disorder. This is how ischemic chest pain (angina pectoris), heart attack, cerebral circulation disorder or cerebral infarction evolves. The intestinal system’s vascular disease leads to ischemic pain in the bowels with yet no morphological alterations and emerging 10-15 minutes after meals (abdominal angina) or erosive-ulcerative ischemic colitis which comes with tissue damage. If this circulatory disorder becomes complete (e.g. mesenterial thrombosis), it might lead to definitive intestinal infarction. Another example for ischemic pain is periodic walking ability due to circulatory limitations of the lower limbs, that is intermittent claudication (pain emerges in the tights or shin during walking, and making the patient to stop).
Severe vascular stenosis might lead to limb necrosis or gangrene (foot gangrene). Leriche’s syndrome is a special form: aortoiliac stenosis with impotence. Affection of renal vessels leads to renovascular arteriosclerosis and secondary hypertension. Abnormal (turbulent) flow around the stenosis (e.g. at the carotid artery bifurcation) are heard as murmurs with auscultation. The pulse might get weaker after the stenosis.
I/2.3.7.2.10: A special and rare form of arterial hardening: Mönckeberg’s medial calcific sclerosis
Typically diffuse or focal calcification occurs in the middle layer of medium size arteries of the shin. Primarily diabetic patients are affected above 50 years of age. Due to calcification, the lesions can be detected radiographically.
|
|