 |
VIII./6. fejezet: Mérési adatok értelmezése: oki kapcsolat keresése a tünetek és a variánsok között
Bevezetés
A ritka örökletes betegségekben szenvedők gyakran 5-30 évet is várnak a diagnózis megszületésére, az átlagos diagnosztkai késedelem 7 év. Azon érintettek körében, ahol a végül kóroki genetikai eltérés igazolást nyer, 40%-ban a kezdeti diagnózis helytelennek bizonyul. Egy WES vizsgálat esetén 25-100 ezer variáns kerül meghatározásra a szekvenálási mélységtől és a célrégiók feldúsítására alkalmazott módszer függvényében. A WGS esetében kb. 5 millió. Nem indokolatlanul hasonlítják gyakran a WES / WGS adatok bioinformatikai elemzését egy tű szénakazalban történő kereséséhez.
A tapasztalatok szerint minél gyengébben körvonalazható a betegség, annál nehezebb az oksági kapcsolat felderítése a genotípus és a fenotípus között. Minél kevésbé specifikált a diagnózis, annál átfogóbb keresésre van szükség.
Miközben a szekvenáló mérési platformok, referenciaadatok, szoftverek és elemzési stratégiák jelentősen javultak az NGS technológia első klinikai célú bevetése óta, a releváns genetikai variáns-jelöltek kiválasztására, sorrendezése vonatkozó irányelvek még mindig gyerekcipőben járnak. A WES- és a WGS-alapú kísérletek nagyszámú variánst (ti. referencia szekvenciától való eltérést) azonosítanak, amelyek túlnyomó többsége a vizsgált betegség kontextusában egyáltalán nem releváns, és nagyon valószínű, hogy funkcionális hatása sincs az érintett fehérjére és/vagy szisztémás szintre. Egyelőre nem létezik „univerzális recept" a megoldásra, amely használható lenne a funkcionális variánsok azonosítására.
|
 |
VIII./6.1. Az ismert kóroki variánsok azonosítása
Az ismert jelentőségű variánsokra vonatkozó ismeretek folyamatosan bővülnek. Az American College of Medical Genetics and Genomics (ACMG) kezdeményezésére klinikai laboratóriumi vezetőkből és klinikusokból álló munkacsoport kidolgozott továbbá egy olyan eljárást, amely feltételezett mendeli betegség összefüggő génekben azonosított variánsok osztályozását teszi lehetővé a következő öt kategóriába:
-
1. kóroki,
-
2. valószínűleg kóroki,
-
3. ismeretlen jelentőségű,
-
4. valószínűleg jóindulatú és
-
5. jóindulatú variánsok.
A megalkotott klasszifikációs szabályoknak a célja annak megállapítása, hogy egy mendeli betegségben meghatározott szerepű génben található variáns állhat-e az adott betegség hátterében. A variánsok klasszifikációját kritériumok két halmaza teszi lehetővé: egyiket a patogén és a valószínűleg patogén variánsok, másikat a jóindulatú és valószínűleg jóindulatú variánsok besorolásáért hozták létre. Minden kritérium súlyozott, így vannak nagyon erős, erős, mérsékelt, vagy támogató patogenitási kritériumok; illetve önálló, erős vagy támogató jóindulatú viselkedésre utaló kritériumok.
A kritériumokat ezt követően a pontozási szabályoknak megfelelően kombinálják, hogy az adott variáns az ötszintű rendszer egyik kategóriájába kerüljön. Abban az esetben, ha egy variáns a két halmaz egyik kritériumának sem tesz eleget, illetve ha a bizonyíték jóindulatú vagy kóroki jellegét tekintve ellentmondásra vezet, akkor a variáns bizonytalan jelentőségűként értelmezhető.
|
 |
VIII./6.2. Az ismeretlen variánsok jelentőségét jósló in silico megoldások
Amennyiben a vizsgált egyén klinikai tüneteinek jól megfeleltethető betegséget, előzetesen összefüggésbe hozott patogén vagy valószínűleg patogén variáns(oka)t sikerül azonosítani, akkor további lépések nem is szükségesek az értelmezés ezen szakaszában. Azonban bizonytalanság vagy ismert variáns-jelöltek adatok hiányában tovább kell lépnünk, a nem karakterizált nagyszámú variánsról kell eldönteni, hogy összefüggésben állhat-e az adott variáns a megfigyelt kórképpel vagy sem.
Számos in silico eszköz segítheti a szekvencia-variánsok, elsősorban a misszensz és az intron-exon határon elhelyezkedő splice-site variánsok interpretációját. Az egyes eszközök által használt algoritmusok eltérhetnek, de képesek meghatározni a variáns hatását a nukleotid és aminosav szinten, beleértve a variáns hatásának megállapítását az elsődleges és alternatív gén transzkriptumokra, és így a fehérjére is. A predikciók figyelembe veszik a pozíció evolúciós konzerváltságát és a korábban patogénként leírt eltérések jellemzőit is, utóbbit a gépi tanulás tanító halmazaként használják fel. Mivel ezek a programok csupán előrejelzést készítenek, ezért nem lehetnek kizárólagos forrásai a patogenitás bizonyítékának egy klinikai értékelésben.
|
|
VIII./6.3. Variánsok tudásvezérelt szűrése
Még a megfelelő kísérleti tervezés és a hatékony szűrési protokollok mellett a WGS és WES vizsgálatok gyakran több, valószínűleg funkcionális következménnyel járó variánst jelölnek meg, mint amennyinek a kísérletes igazolására lehetőség van. A fehérje-kódoló régióban elhelyezkedő variánsok fehérjére gyakorolt hatása jól előrejelezhető, ez alapján történő rangsoroláshoz képest a legerősebb variánsjelöltek kiválasztása egy adott betegség vagy fenotípus esetében már nem mindig nyilvánvaló. A rendelkezésre álló biomedikai ismeretekkel és funkcionális variánsokkal rendelkező génjelöltek megítélése fontos lépés a még kezelhető számú variáns-jelöltek a további validálási vagy funkcionális jelentőségének feltárása érdekében végzett kísérletek előtt. Számos tényezőt kell figyelembe venni ezen a ponton:
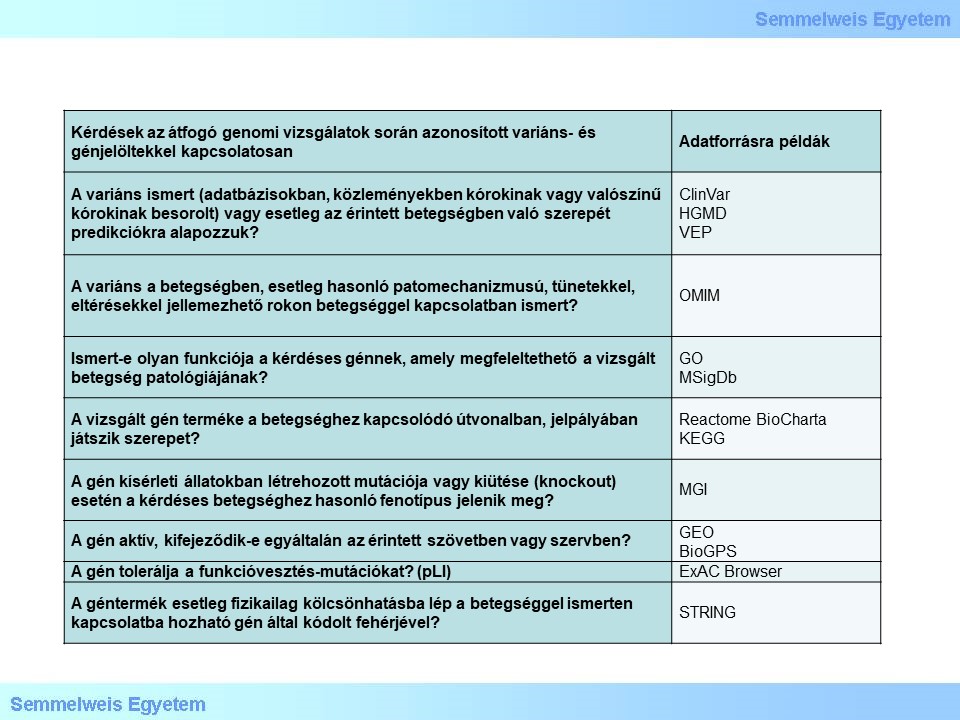
2.táblázat Ismeretlen jelentőségű variáns és betegséggén-jelöltek karakterizálása
|
VIII./6.4. Virtuális panelvizsgálat
A genomikai szinten hívott variáns-tömeg kezelésében egy egyik lehetséges megközelítés, ha a megfigyelt klinikai képet a vezető tünetek, laboreltérések stb. alapján besoroljuk egy betegségcsoportba, majd a kapcsolódó, ismert génekre korlátozva próbáljuk meg az állapotért felelős eltérést azonosítani.
Jóllehet a jelölt génekre támaszkodva könnyebben átfoghatóvá válik a mérlegelésre alkalmas variánsok száma, az adott állapottal az eddigiekben még összefüggésbe nem hozott gének a látókörön kívül maradnak, valamint elveszítjük azokat a farmakogenetikai, prediktív genetikai adatokat is, melyek a vizsgált egyén szempontjából komoly egészségnyereséggel járhatnának.
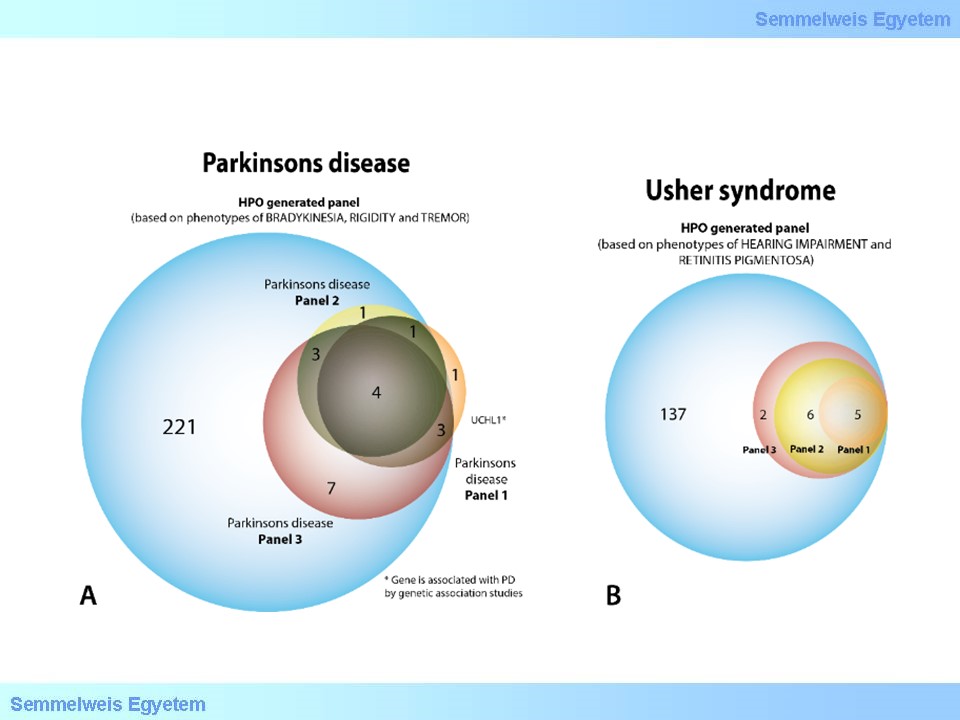
7.ábra: Alternatív megközelítést jelent a variánsok megfeleltetése ismert, betegséggel összefüggő génszettekre, virtuális panelekre történő szűréssel.
|
Amennyiben a fellelt variáns egy olyan génben található, amelyet még nem hoztak kapcsolatba az adott fenotípussal, úgy a bizonytalan jelentőségű génben található („gene of uncertain significance”, GUS) és persze ekkor a variáns-jelölt is bizonytalan jelentőségűnek tekinthető.
|
|
VIII./6.5. Öröklésmenet és zigozitás
A variánsok jelentőségének egyéni mérlegelése során az adott génről, illetve a betegség-asszociációiról rendelkezésre álló adatok mellett mérlegelendő szempont persze az is, hogy
- A feltételezett fenotípusra jellemző öröklésmenet összeegyeztethető-e a kérdéses variáns alléljainak számával a vizsgált egyénben. Pl. ha történetesen autoszómális recesszív öröklésmenettel jellemezhető génben vagyis amikor egy gén funkcióvesztését okozó mutáció(i/k)nak mindkét, anyai és apai lókuszon is meg kell jelenniük a betegség tüneteinek manifesztálódásához, de azt egy heterozigóta formában találjuk meg, az akár ismert patogén variáns esetén sem lehet elégséges a megfigyelt fenotípusunk magyarázataként.
- A családon belüli szegregáció, amennyiben több családtagról is rendelkezünk genomi adattal, az érintettekben való kizárólagos előfordulás egy ritka variáns esetén erős bizonyítékot szolgáltat. Ritka funkcionális következménnyel járó de novo variánsok előfordulása egy betegséghez kapcsolható génben szintén erősen felveti a betegség-okozó képesség lehetőségét.

8.ábra: Elemzési stratégiák több minta felhasználásával. A különböző esetekben a cél, hogy a) érintettekben igen, a tünetmentesekben nem kimutatható, b) tünetmentes szülők rokonházassága esetén a homozigóta biallélikus variánsokat, vagy c) a valamelyik szülőtől örökölt heterozigótaság elvesztését (LOH: loss of heterozygosity), d) a hasonló fenotípusú családok érintett tagjait vizsgálva a közös, esetleg e) a tünetmentes biológiai szülőkben teljesen hiányzó, de novo keletkezett variánsokat azonosítsuk. Végül f) a kórfolyamatban szerepet játszó ismert vagy feltételezett génekre korlátozva is megkísérelhető a genetikai háttér megfejtése.
|
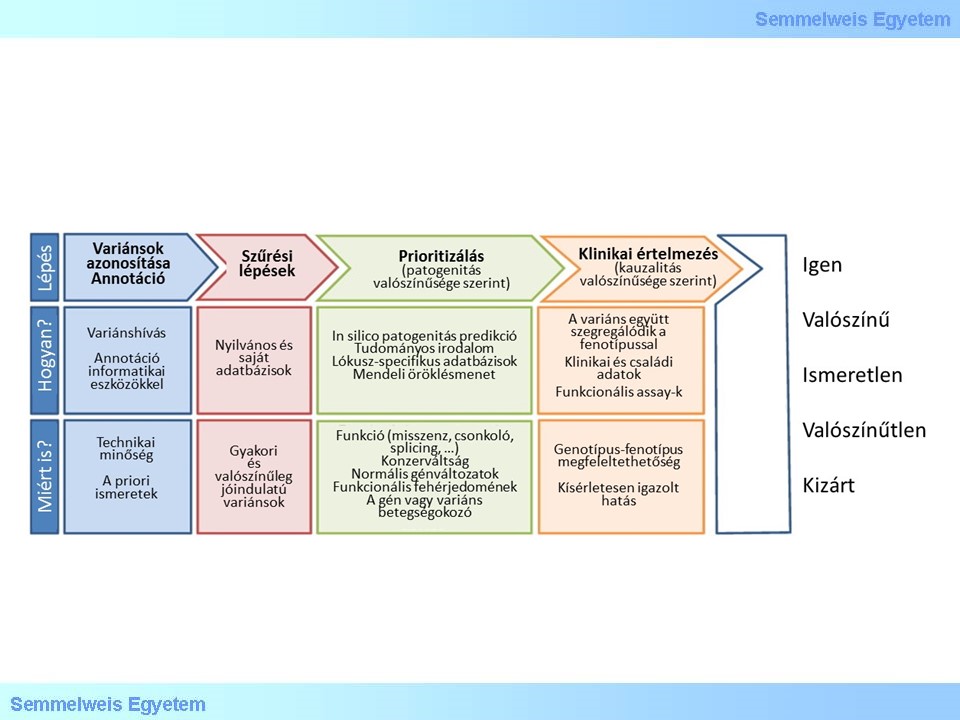
9.ábra: Elemzési stratégiák, célok és lehetőségek lépésekre tagolva (forrás)
|
Ha nem sikerül meggyőző jelöltet kiemelnünk, ezt követően a szűrés helyett talán szerencsésebb variánsprioritizálásról, vagyis a variánsok és az általuk befolyásolt gének relevancia szerinti rangsorolásáról beszélni, mert nem definiálhatók szigorú vagy általánosan kalibrálható határok és súlyok a leíró változók sokdimenziós terében.
VIII./6.6. Variánspriotizálók
A nagyszámú detektált alternatív allél között történő kereséshez gyakran
szükséges egy prioritási lista felállítása aszerint, hogy mely variánsok esetében lehet nagyobb valószínűséggel egy lehetséges kóroki szerepével számolni. A számos különböző szempontot figyelembe venni képes szoftverek, a variánsprioritizáló algoritmusok
-
- jellemzően az eltérés típusából levezethető, a fehérje funkciójával interferáló hatás mértéke,
-
- az érintett gén funkcióvesztése esetén megfigyelhető ismert fenotípusnak a kérdéses klinikai képhez való megfelelősége, valamint
-
- a várt és megfigyelt öröklésmenet
együttes figyelembe vételével képesek egy sorrendet felállítani.
Gépi olvasásra alkalmas módon írható le az egyén fenotípusa egy, a humán rendellenességeket katalogizáló ontológia (pl. HPO, Human Phenotype Ontology) segítségével.

10.ábra: Variánspioritizálás
|
|
|