 |
XVI./ 3.3.4. Szekvenálási metodikák
XVI./ 3.3.4.1. Sanger szekvenálás
A Sanger-féle (dideoxi vagy láncterminációs) szekvenálás során a DNS-ben található normál nukleotidok (dNTP) mellett didezoxinukleotidok (ddNTP) is jelen vannak. A didezoxinukleotidok abban különböznek a normál nukleotidoktól, hogy a 3’ szénatomon hidroxil-csoport helyett hidrogén található, így amikor ezek beépülnek a DNS láncba a további szintézist megakadályozzák. A dideoxi-nukleotidok mennyisége 1%-a a normál nukleotidok mennyiségének. Manapság 4 különböző florokrómmal jelölt ddNTP-t használunk, amelyeket a reakció végén a kapilláris elektroforézis segítségével valasztunk el. A kapott nyers szekvenciát egy analizáló program alkalmassá teszi a további analízisre. FASTA formátum lehetővé teszi a referencia genomhoz való könnyű illesztést.

1. ábra: Sanger szekvenálás elve
|
XVI./ 3.3.4.2. Piroszekvenálás
A piroszekvenálás során a DNS másolásakor beépülő nukleotidról leszakad egy pirofoszfát. Ezt a reakcióban egy luciferáz enzim által okozott fény felvillanása jelzi.
|
|
A piroszekvenálás a „szekvenálás szintézissel”' elvén alapul, vagyis egy egyszálú DNS templátról enzimatikusan komplementer szálat szintetizálnak. Valós időben a DNS- polimeráz aktivitását detektálják egy kemilumineszcens enzim segítségével. Amikor egy nukleotid beépül a DNS szálba, pirofoszfát (PPi) keletkezik, és ennek a mennyiségét mérjük egy kapcsolt reakcióval, ami végső soron luciferin/luciferáz rendszer felvillanásával jelez. Egyszerre csak egyféle nukleotidot adnak a rendszerhez, így biztos, hogy csak egy nukleotid fog beépülni a növekvő szálba. Ha több nukleotid épül be, akkor arra a fényintenzitás növekedéséből lehet következtetni. Kemilumineszcencia csak a megfelelő, komplementer nukleotid esetében következik be, hiszen csak akkor szabadulhat fel a pirofoszfát. A nem komplementer, felhasználatlan nukleotid, még a következő bázis beépülése előtt degradálódik. Alapvetően azt kell detektálni, hogy éppen milyen nukleozid-trifoszfátot adtak a rendszerhez, és az beépült-e az egyszálú DNS-láncba vagy sem.
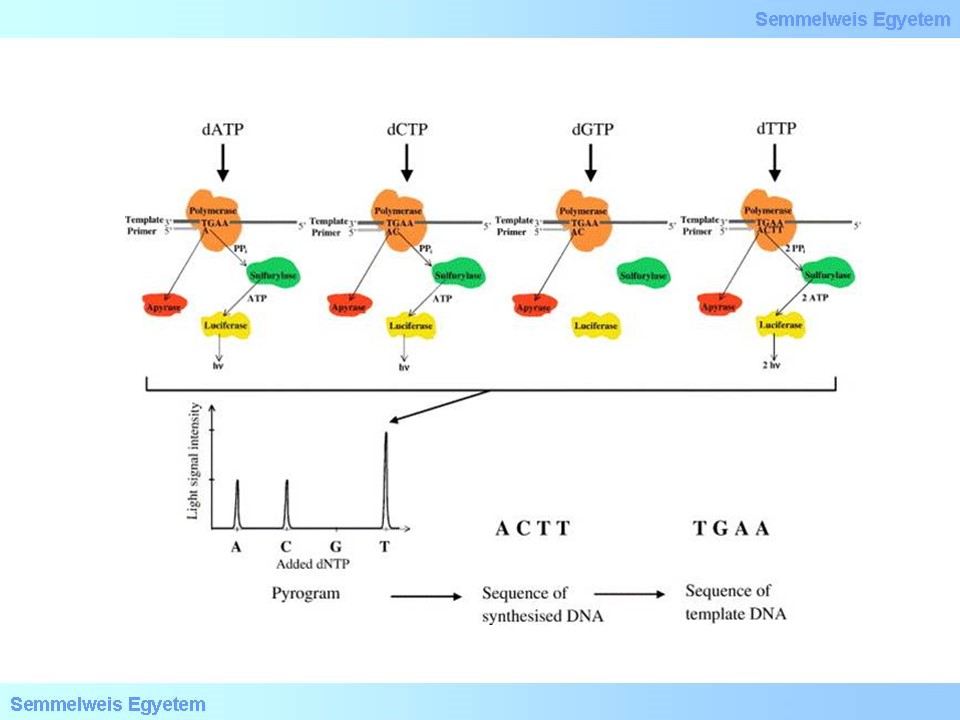
2. ábra: Piroszekvenálás elve (https://www.semanticscholar.org/paper/Pyrosequencing)
|
Rutindiagnosztikában jól alkalmazható:
-
- Mutációk pontos mozaicizmus rátájának a megadása (pl. tumorokban szomatikus mutációk vizsgálata – KRAS 2.exon 12. és 13. kodon),
-
- Mitokondriális DNS pontos heteroplazmia arányának meghatározása.
|
|
XVI./ 3.3.4.3. Újgenerációs szekvenálás (NGS – Next Generation Sequencing)
Az NGS párhuzamosan sok minta nagy áteresztőképességgel (deep sequencing) történő vizsgálatának lehetőségét teremti meg. Az új-generációs módszerek mind a láncszintézis, mind a detektálás terén a hagyományos Sanger-féle szekvenálástól lényegesen eltérő technológiát használnak. Kivitelezéséhez fejlett informatika és robottechnika szükséges. A szekvenálás során viszonylag rövidebb, néhány 100 bázispáros DNS szakaszokat párhuzamosan sok kópiában (akár 1 millió) szekvenálunk.A módszernek köszönhetően a DNS-alapú vizsgálatok hatékonysága, átfutási ideje és a fajlagos költsége csökkent.
-
- Teljes genom (WGS – whole genome sequencing),
-
- Teljes exom (WES – whole exome sequencing),
-
- Teljes transzkriptom (RNAseq),
-
- kis RNS-ek (small RNA-Seq),
-
- CHipseq szekvenálásra (ennél a módszernél az ún. kromatin immunoprecipitációval előállított génexpresszió szabályozásban résztvevő DNS régiókat szekvenálják új-generációs módszerekkel),
-
- NGS panelek (target szekvenálás) - célzottan, betegségre specifikus gének egyidejű szekvenálása.
|
|
NGS metodikai részfolyamatai
-
Könyvtárkészítés,
-
Klonális amplifikáció
-
Bioinformatikai kiértékelés
Klonális amplifikációBioinformatikai kiértékelés
A szekvenálni kívánt DNS-t (akár teljes genomot) apró darabokra fragmentálják (<300bp) és barcode (index szekvenciák) hozzáadásaval biztosítják a minták azonosítását. A dupla szálú DNS két végét kijavítják (ragadós végek eltüntetése) 3’ végeket adenilálják és ehhez egy timin túlnyúló véggel rendelkező adapter DNS-t ligálnak, majd a ligált DNS-darabokat szelektálják, és egyszálúsítják NaOH-dal.
Klonális amplifikáció - Szekvenálás
Jelenleg az egyik legáltalánosabb módszer az Illumina/Solexa szekvenálás, amely a „Híd-amplifikáció módszert” használja. Ennek során a mintát egy szekvenáló lemezre viszik, amelyre az adapterrel komplementer oligonukleotidokat (primerek) horgonyoznak ki ahová a DNS fragmentumok az egyes szálak között klasztereket képezve kötődnek. Az amplifikáció állandó hőmérsékleten (60°C) megfelelő puffer és denaturálószer jelenlétében megy végbe. A reagensek minden egyes ciklus után lemosódnak. A PCR reakció végén az eredetileg a horgonyra hibridizált dupla szálú DNS templát szálát formamiddal leválasztják és lemossák. Az egyszálú DNS elhajlik, és a szabad végével egy másik lehorgonyzott primerrel hibridizál. Ezzel is végbemegy a PCR, és a végső denaturáció. Ezt követően az átírt szálak egymással vagy másik primerrel hibridizálhatnak és duplikálódnak, majd további amplifikáció után készen állnak a szekvenálásra.

3. ábra: Bridge-amplifikáció folyamatábrája (Rizzi et al. Genet Sel Evol. 2012 Jul 6;44:21)
|
Szekvenálás során a templáthoz egy primert ligálnak, majd a reakcióhoz egyszerre adják hozzá a 4 nukleotidot tartalmazó különbözö fluoreszcens festékkel rendelkező reverzibilis terminátorokat.

4. ábra: Egy reverzibilis nukleotid analóg szerkezete (elte.prompt.hu)
|
A beépült nukleotid fluoreszcens jelet ad, amely CCD kamerával detektálható. A nukleotid beépülése után a flurofort és a 3' blokkolót kémiai úton levágják, majd lemossák a be nem épült nukleotidokkal együtt.
A szekvenálás idejét a vizsgálat célja határozza meg. Egy teljes genom szekvenálása átlagosan 7-9 napot is igénybe vehet.
Bioinformatikai értékelés
A szekvenálást követően a szekvencia szakaszokat (read-eket) a genomösszerakás során a leolvasásokat összeillesztjük az átfedő részszekvenciák alapján nagyobb, összefüggő DNS szakaszokká, ún. kontigokká. Ezt követően lokális szekvenciaillesztéssel egy adott szekvenciához hasonló szekvenciákat keresünk egy annotált referencia-adatbázisban. Majd a folyamat végén a génfelismerés és funkcionális annotáció (gene calling) történik, amely során az összerakott kontigokhoz hasonló géneket rendelünk.

5. ábra: NGS bioinformatikai kiértékelésének összefoglalása (elte.prompt.hu) (Doitsidou et al., Genetics. 2016 Oct;204(2):451-474.)
|
Biológiai értékelés
A klinikai gyakorlatban sokszor annak eldöntése, hogy melyik variáns áll a beteg tünetei mögött korántsem egyértelmű. Ennél a folyamatnál több stratégiát követhetünk, mint a fenotipizáláson alapuló módszerek, adott betegség hátterében ismert gének/génpanelek célzott szűrése, biológiai funkciók, jelátviteli útvonalak alapján. A variánsok szűrésénél általában a ritka variánsokra (MAF<1%) fókuszálunk, így első körben a populációs adatbázisok frekvencia adatait vesszük figyelembe, mint pl. az 1000 genom projekt és gnomAD (Genome Aggregation Database) adatbázisok. Fontos az, hogy az adott populációra specifikus értékekkel számoljunk, így egy kaukázusi beteg esetében a kaukázusi populációra jellemző minor allél értékekkel (MAF) számoljunk.
Szerencsés esetben olyan eltérést találunk, amely az adott betegség hátterében korábban leírt patogén mutáció. Amennyiben ilyen eltérést nem találunk az adott variáns evolúciós konzerváltsága vagy általa okozott konformáció változást jósló in silico predikciós algoritmusok alkalmazásával tudunk az eltérés patogenitására következtetni, majd célzottan családi szegregciós vizsgálatot végezni. A variánsok jelentőségének pontos eldöntése funkcionális sejtbiológiai mérésekkel kivitelezhető, bár ez a gyakorlatban ennek magas költség és időhányada miatt nagyon minimális esetben történik. A variánsok végső besorolásában a ACMG (American College of Medical Genetics and Genomics) irányelve nyújt hasznos segítséget.
|
|