|
V./5.2. Lysosomalis tárolási betegségek
A lysosomalis tárolási betegségek a lysosomalis enzimek/fehérjék nem megfelelő működése folytán alakulnak ki, melyek hátterében öröklődő génhiba áll. A lysosomák betegségeinek jelenleg körülbelül 50 ismert típusa létezik. Általánosságban autoszomális recesszív öröklésmenetűek, a Fabry-kór és a Hunter-szindróma kivételével, melyek X-kromoszómához kötött recesszív öröklésmenetet követnek. Az autophagocytosis kapcsán keletkezett, vagy az extracelluláris eredetű emésztetlen makromolekulák felhalmozódása okozza a diverz tüneteket. A lysosomákban felhalmozódó szubsztrátok szerint megkülönböztetünk sphingolipidosisokat, mucopolysaccharidosisokat, glikoproteinszisokat, lipidosisokat, glikogenosisokat és oligoszacharidosisokat. Ennek megfelelően tárgyaljuk jelen fejezetben a lysosomális betegségeket. A megértéshez feltétlenül szükséges, hogy felelevenítsük a membránalkotó lipidek (11.ábra) és ezeken belül a szfingolipidek főbb csoportjait (12.ábra). A foszfolipidek kétféle vázon alakulnak ki, a glicerinen (glicerofoszfolipidek) és a szerkezetileg hasonló szfingozinon (szfingo-foszfolipidek).
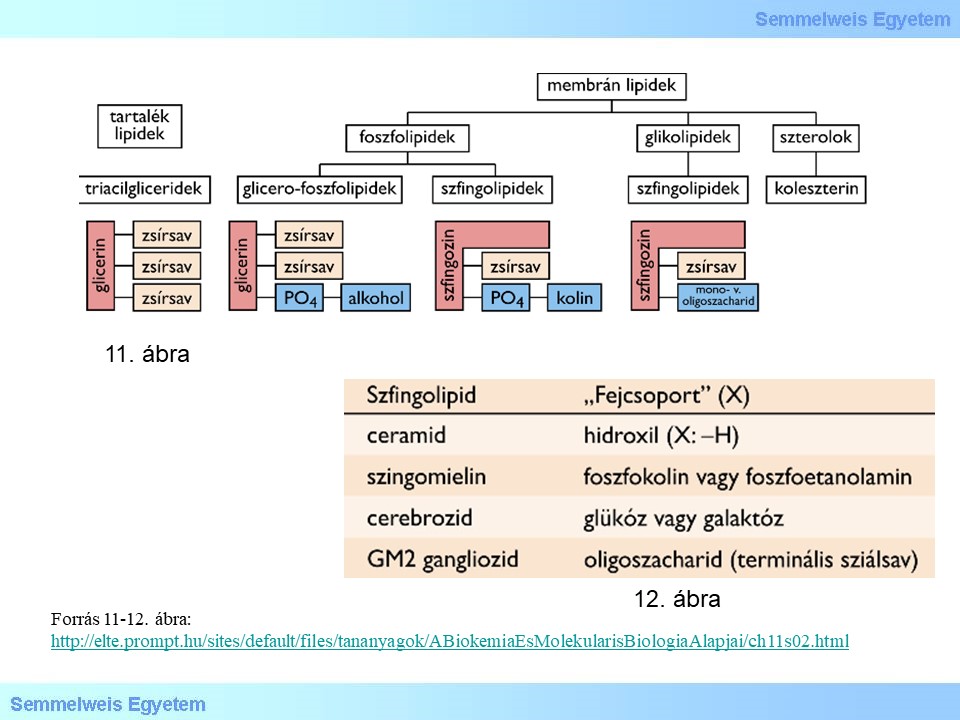
11. ábra: A membránalkotó szerkezeti lipidek típusai és összetétele (a trigliceriddel összevetve)
12. ábra: A különböző sphingolipidek neve és összetevői
|
V./5.2.1. Sphingolipidtárolási betegségek és lipidosisok

3. táblázat: A legfontosabb sphingolipidosisok klinikai tüneteinek összefoglalása
|
|
|
V./5.2.1.1. Tay-Sachs-kór
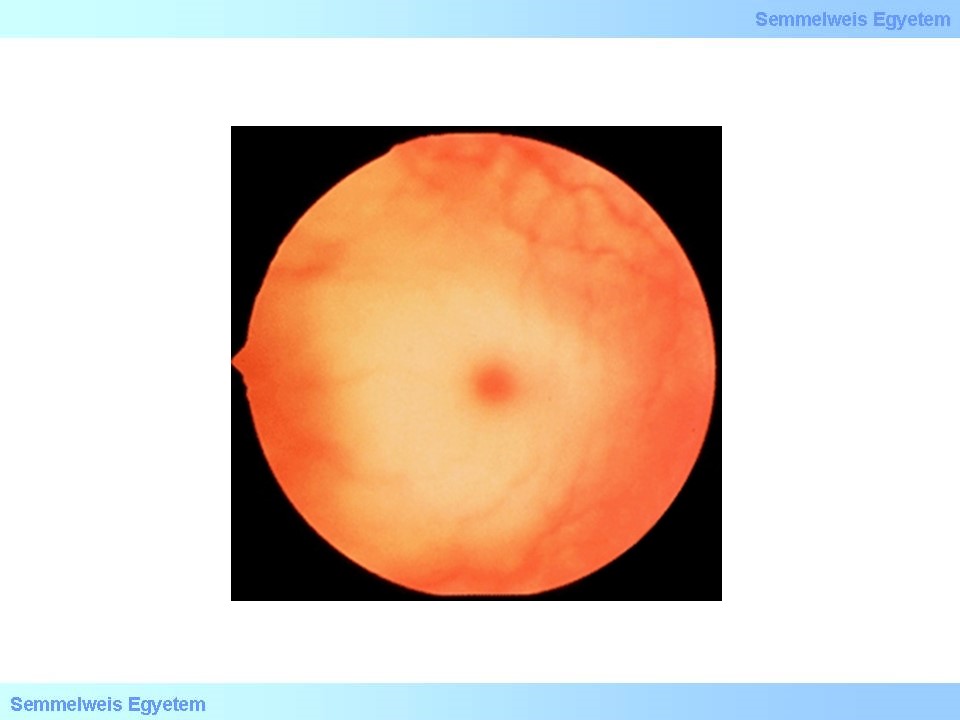
A Tay-Sachs-betegség tulajdonképpen egy GM2-gangliosidosis, tehát GM2 gangliosidok és származékai halmozódnak fel a hexózaminidáz A enzim aktivitásának csökkenése vagy teljes kiesése miatt. A felhalmozódó anyagok a neuronok fokozatos pusztulását idézik elő az agyban és gerincvelőben. Leggyakoribb formája már gyermekkorban tüneteket okoz. A betegségben szenvedő csecsemők általában 3-6 hónapos életkorig normálisan fejlődnek, ezt követően viharos gyorsasággal romlanak motoros készségeik. A betegség előrehaladtával rohamok, látás és halláskárosodás, súlyos értelmi fogyatékosság és teljes bénulás alakul ki. A progresszív látásromlás a n. opticus atrophiája miatt alakul ki. A szemészeti vizsgálatok kapcsán derül fény a maculán egy cseresznyepiros folt jelenlétére az esetek 90%-ban.
|
A Tay-Sachs-betegség infantilis formájában szenvedő gyermekek általában csak pár éves korukig élnek. Tay-Sachs-betegség egyéb formái nagyon ritkák, gyermekkorban, serdülőkorban vagy fiatal felnőttkorban jelentkezhet a betegség, és általában enyhébb tünetekkel jár, mint az infantilis forma. Az izomgyengeség, ataxia, mentalis retardatio ezekben a formákban is jellemző.
|
|
|
V./5.2.1.2. Niemann-Pick-betegségcsoport
A Niemann-Pick betegségcsoport 3 altípusát különíthetjük el, az A és B a sphingolipidosisokhoz, a C a lipidosisokhoz sorolható. Bővebben a Niemann- Pick-C típusával foglalkozunk. Az NPC1 és 2 lizoszomális fehérjéket (nem enzimek!) kódoló megegyező elnevezésű gének defektusa az intracelluláris koleszterinek és koleszterin-észterek transzportzavarát és felhalmozódását okozza. A genetikai kiváltó eltérés alapján elkülöníthetünk C1 és C2 típusú Niemann-Pick betegséget, bár a tüneteik nagyon hasonlóak. A betegség általában már gyermekkorban nyilvánvalóvá válik (első tünet általában az icterus), de ismertek későbbi indulású formák is. Vezető tünet az ataxia, a supranuclearis tekintésbénulás, görcsök, hepatosplanomegalia, interstitialis tüdőbetegség. Progresszív mentalis hanyatlással jár a C1 és C2 típusú Niemann-Pick betegség is, gyakori a beszéd- és a nyelészavar, amely idővel romlik. A mutáció következményeinek súlyosságától függően az érintettek megérhetik a felnőttkort.
Ebben a videoban nagyon jól szemléltetik a betegség tüneteit:
|

Nagyon ritkán fordul elő az ún. késői indulású forma (II-es típus), amelyben a tünetek széles határok között változnak és jó pár év túlélés jellemző a diagnózist követően.
|
V./5.2.1.3. Krabbe-betegség
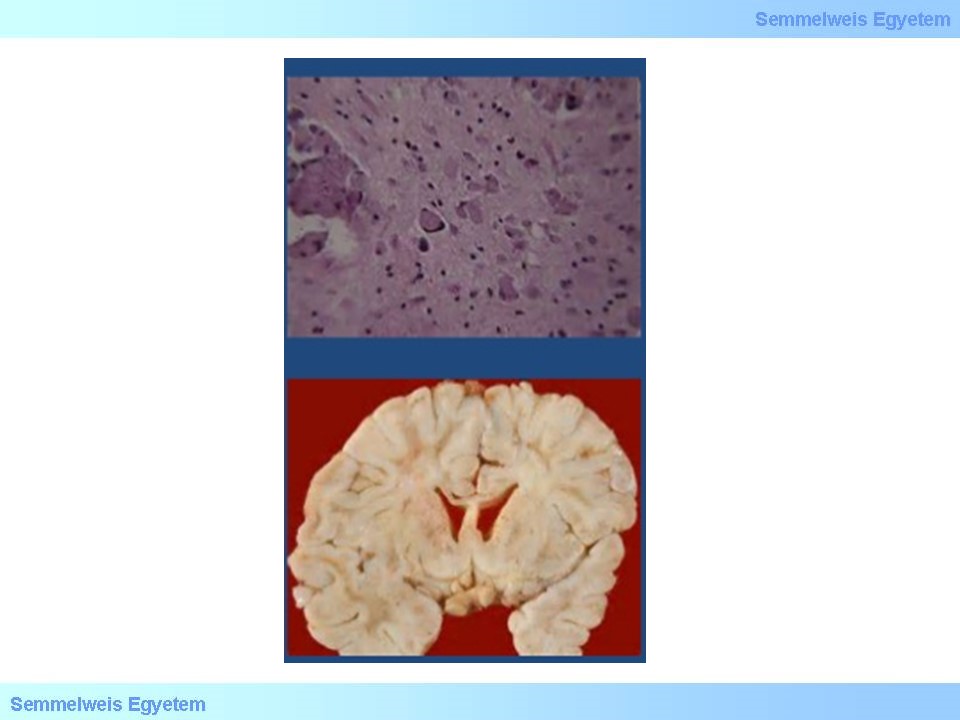
A Krabbe-betegség leggyakoribb formájában, az I-es típusban érintett újszülöttek az első pár életévükben elhaláloznak. Nagyon súlyos mentalis retardatio, motoros funkciózavar, perifériás neuropathia, vakság és süketség uralják a kórképet. A galaktocerebrozidáz enzim defektusaként alakul ki progresszív leukodystrophia, mely a fenti tüneteket okozza. A kórképet szokás még globoid sejtes leukodystrophiaként is említeni, utalva a jellegzetes PAS pozitívan festődő ún. globoid sejtekre, melyeket a 14. ábrán figyelhetsz meg.
V./5.2.1.4. Gaucher-kór
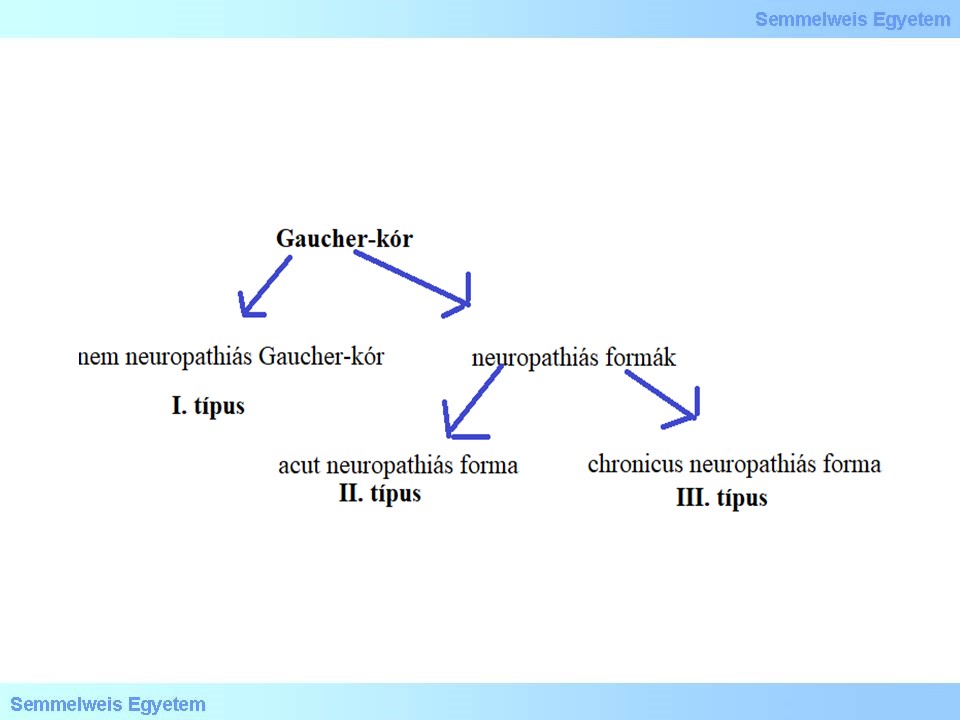
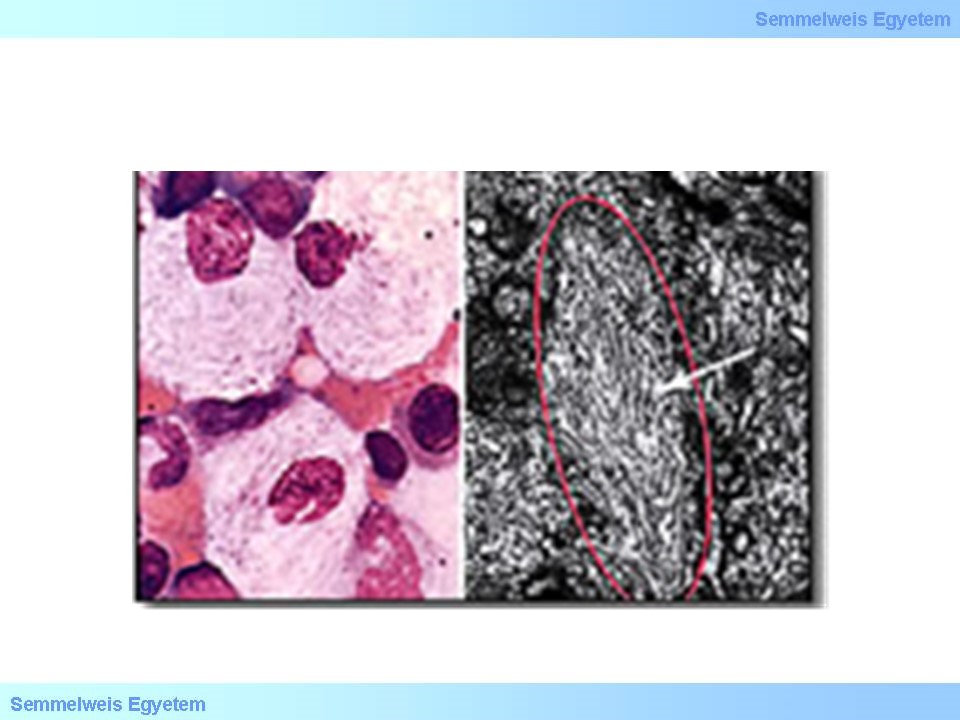
A glucocerebrosidok lebontása szenved zavart a Gaucher-kórban, mely rendkívül heterogén tünetekkel jár, így klasszifikációja nehéz, ld. 15. ábra. Az I. típusú, ún. nem neuropathiás Gaucher-kór a leggyakoribb formája ennek a betegségnek. Ennek a betegségnek a jellemzői enyhétől a súlyosig terjednek, és gyermekkortól felnőttkorig bármikor megjelenhetnek. A fő tünetek közé tartoznak a hepatosplenomegalia, anaemia, thrombocytopenia, leukopenia (fokozott infectio hajlam) csontok rendellenességei (csontfájdalom, törések) és ízületi gyulladás. A betegség lényegesen nem rövidíti meg a várható élettartamot. A felhalmozódó anyagok jellegzetes szövettani képet idéznek elő, ld. 16. ábra.
A II. és III. típusú Gaucher-betegséget az ún. neuropathiás formáinak nevezik, mivel azokat a központi idegrendszert érintő problémák jellemzik. A fent leírt tünetek mellett görcsöket és súlyos mentalis kárososdást okoznak. A II. típusú Gaucher-betegség acut neuropathiás forma, újszülöttkorban jelentkezik, bulbaris tünetekkel majd teljes dysphagia alakul ki spasticitás, gyakori infekciók mellett. A III. típusú chronicus neuropathiás Gaucher-betegség későbbi indulású, de kamaszkor előtt mindenképp jelentkeznek a nagyon változatos tünetek (tekintészavar itt is lehet, mint az NPC-ben!), progressziójuk jóval lassabb.
.
|
|
|
V./5.2.1.5. Fabry-betegség
A Fabry-betegség nemhez kötött öröklődésű, az érintett férfiak esetében alig találunk alfa-galaktozidáz enzimaktivitást, míg hordozó nőkben az enzim aktivitása változó mértékben csökkent. A szövetekben (az erek simaizom és endothelsejtjeiben) felhalmozódó glikosphingolipid, a globotriaozil ceramid vagy GL-3 okozza a tüneteket. Jellemző tünetei a fájdalmas acroparaesthesiák különösen a kézben és a lábakban; angiokeratomák; a csökkent izzadási képesség (hypohydrózis); cornea opacitás; tinnitus; halláskárosodás; vesetubulusok károsodása miatt veseelégtelenség. A Fabry-betegségben potenciálisan életveszélyes szövődmények is előfordulhatnak, mint például szívroham, stroke. Néhány érintett esetében a rendellenesség enyhébb formái figyelhetőek meg, amelyek későbbi életkorban jelentkeznek, és csak a szívre vagy a vesére lokalizálódnak.
V./5.2.2. Mucopolysaccharidosisok
A mucopolysaccharidosisok (MPS) esetében a glükózaminoglikánok (GAG) lebontása károsodik. A GAG-ok olyan poliszacharidok, amelyek aminocukrokat, pl. glükózamint tartalmaznak, korábban mucopolysaccharidokként nevezték őket.
A glükózaminoglikánok közé tartozik pl. a heparin, kondroitin-szulfát, hialuronán, heparán-szulfát, dermatán-szulfát, keratán-szulfát. Nagyon fontos strukturális elemei a sejtmembránoknak, kötőszöveteknek, így érthető, hogy a mucopolysaccharidosisok olyan tüneteket okoznak, mint pl. alacsonynövés, csontdeformitások, ízületi problémák, organomegalia, corneahomály.
|
Az Aldurazyme az első és egyetlen FDA által jóváhagyott ERT-kezelés, amelyet rekombináns DNS technológiával fejlesztettek ki az MPS I. egyének számára. https://www.aldurazyme.com.
|
V./5.2.2.1. Mucopolysaccharidosis I.
Mucopolysaccharidosis I. betegségcsoport a leggyakoribb mucopolysaccharidosis. Az α-iduronidáz enzim csökkent vagy hiányzó működése lebontatlan glükózaminoglikánok felhalmozódásához vezet, melynek vizeletből történő meghatározás során észlelt mintázata specifikus lehet a betegségre. Az alábbi, klasszikus csoportosítás nem mindig feleltethető meg a klinikumnak, a nehezen elkülöníthető, gyakran átfedő klinikai spektrum miatt. Az MPS I betegségcsoportban korábban kizárólag csontvelőtranszplantáció jelentett egyedüli kezelési formát, napjainkban már itt is alkalmazhatunk ERT-t, hetente adott iv. α-L-iduronidáz infúzió formájában.
V./5.2.2.1.1. Mucopolysaccharidosis I. H (MPS I. H, Hurler-szindróma)
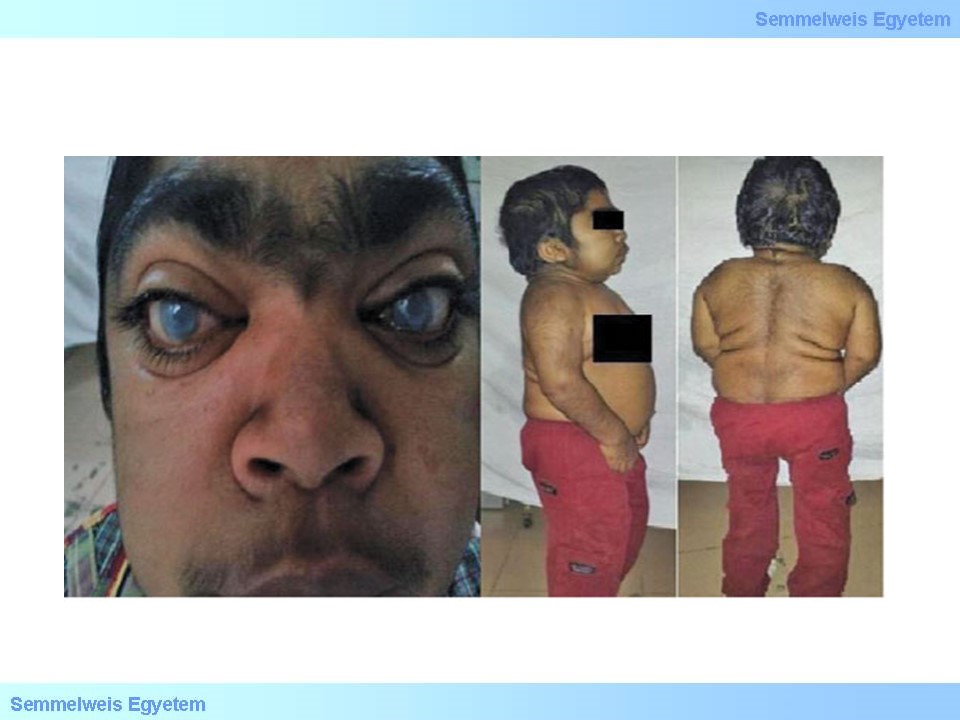
Egyik legsúlyosabb forma, kezelés nélkül korai életévekben halálhoz vezető forma. Az érintett csecsemők féléves kora körül figyelhető meg a jellegzetes, korábban említett, 2. ábrán szereplő vízköpő arcdeformitás, macroglossia, csontdeformitások és hepatosplenomegalia. Súlyos pszichomotoros retardatio, beszédkészség kifejezett zavara, szívbillentyű megbetegedések, cardiomyopathia jellemzik a későbbiekben, illetve a csontdeformitások egyre szembe szökőbbé válnak, kialakulhatnak ún. multiplex disostosisok is. Társulhat még corneahomály, látásvesztés, halláskárosodás. Kezelés nélkül a maximális élettartam kb. 10 év. A korábban említett ERT hatásosságát nagy mértékben korlátozza, hogy nem jut át a vér-agy gáton, így ebben a formában még mindig elengedhetetlen a kezelés szempontjából a csontvelő transzplantáció.
V./5.2.2.1.2. Mucopolysaccharidosis I. H/S (MPS I. H/S, Hurler-Scheie-szindróma)
Az ebben a fejezetben említésre kerülő mucopolysaccharidosisok közepesen súlyos formája. A tünetek kora gyermekkorban kezdődnek, és a mentalis retardatio nagyon enyhe, vagy nem is jellemző. A csontvázrendszeri eltérések, corneahomály, halláskárosodás (esetleg süketség), szívbillentyű megbetegedések kifejezettebbek. A betegek többsége megéri a felnőttkort. Jellegzetes arckarakterisztikával jár, benyomott orrgyökkel, micrognathiával, macroglossiával (17. ábra).
V./5.2.2.1.3. Mucopolysaccharidosis I. S (MPS I. S, Scheie-szindróma)
A legkevésbé súlyos forma a 3 említett mucopolysaccharidosis közül, még későbbi indulású, nem jár jellegzetes arckarakterisztikával vagy csak nagyon enyhével, és az intellektus megkímélt.
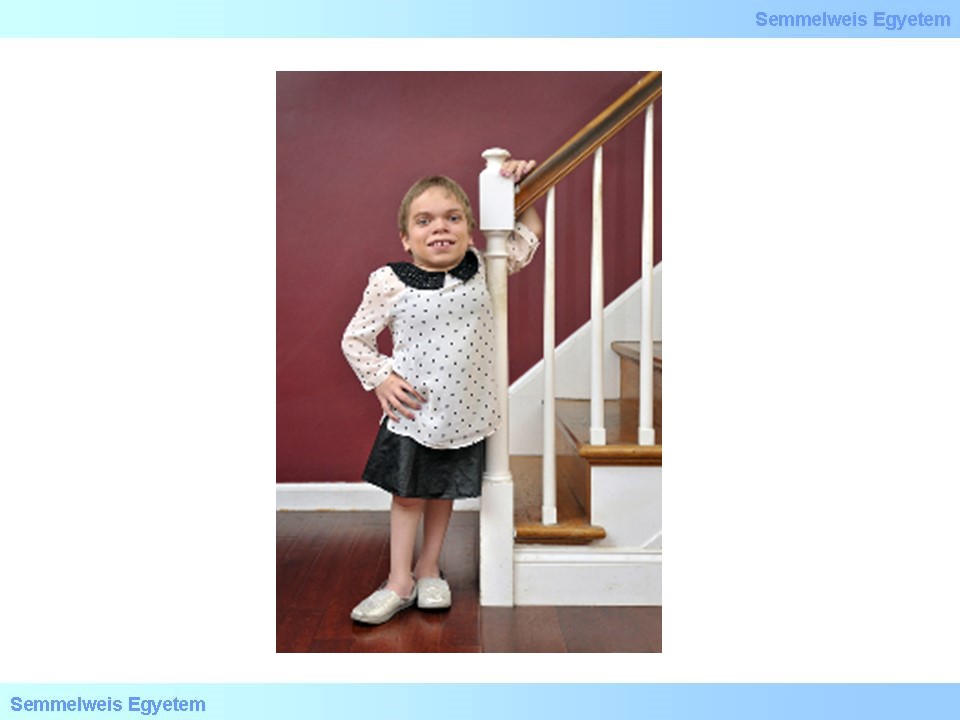
V./5.2.2.2. Mucopolysaccharidosis II. (MPS II, Hunter-szindróma)
Mucopolysaccharidosis II. Hunter-szindrómaként is ismert. Ezt a betegséget az idorunát-2-szulfatáz ezim hibája okozza, a felhalmozódó szubsztrát a heparán-szulfát és a dermatán-szulfát. Öröklődését tekintve X-hez kötött recesszív. Hasonlóan a mucopolysacharidosisok I. típusához, születéskor általában nincs jelen, az első specifikus tünetek (melyek a mucopolysacharidosisok I. típusával nagy mértékben átfednek) pár éves korban jelentkeznek. A corneahomály és a csontvázrendszeri deformitások ebben a csoportban nem jellemzőek, de a jellegzetes vízköpő arc itt is megfigyelhető (18.ábra). A fokozatosan felhalmozódó glycosamynoglycanok miatt a klinikai képhez csatlakozik a cardiomyopathia, légutak obstrukciója, organomegalia, ízületi deformitások, illetve az agy érintettsége miatt a mentális retardatio. Ezen betegség esetében lehetőség van enzimpótló terápia alkalmazására, de újabban CRISPR-Cas9 alapú módszereket is tesztelnek.
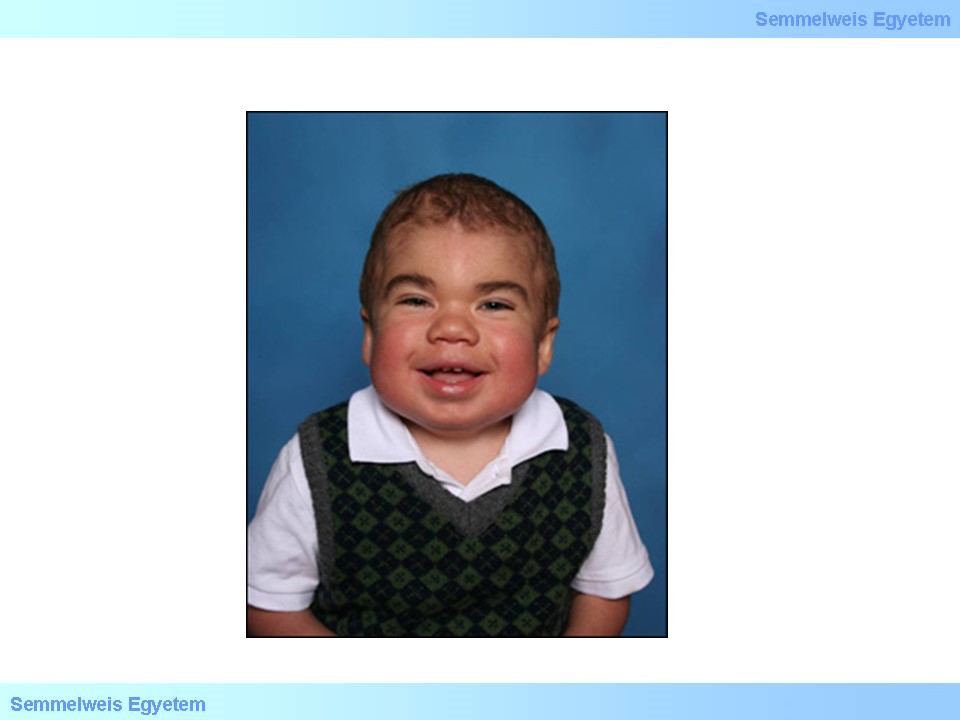
18. ábra: Hunter-szindrómás gyerek jellegzetes arcvonásai, vízköpő arc. Forrás: flipper.diff.org
|
V./5.2.2.3. Mucopolysaccharidosis III. (MPS III., Sanfilippo-betegség)
Több enzim hibája okozza a Sanfilippo-betegséget, a vizeletbe kiválasztott anyag a heparán-szulfát, ez diagnosztikai szempontból is fontos. Elősorban az agyat és gerincvelőt érintő súlyos, progresszív betegség mely pár éves korban ad először tüneteket, melyek főként megkésett beszédfejlődés, viselkedészavarok, agresszió, alvászavarok. Jellegzetes, durva arckarakterisztikával jár, ún. „coarse face” -el. Megfigyelhető még hirsutismus, synophris, vastagszálú, durva haj. Az érintett egyének esetében korai dementálódást figyeltek meg ill. halláskárosodásuk életkoruk előrehaladtával teljes hallásvesztéshez is vezethet. Általában középkorukban haláloznak el.
V./5.2.2.4. Mucopolysaccharidosis IV. (MPS IV., Morquio-betegség)
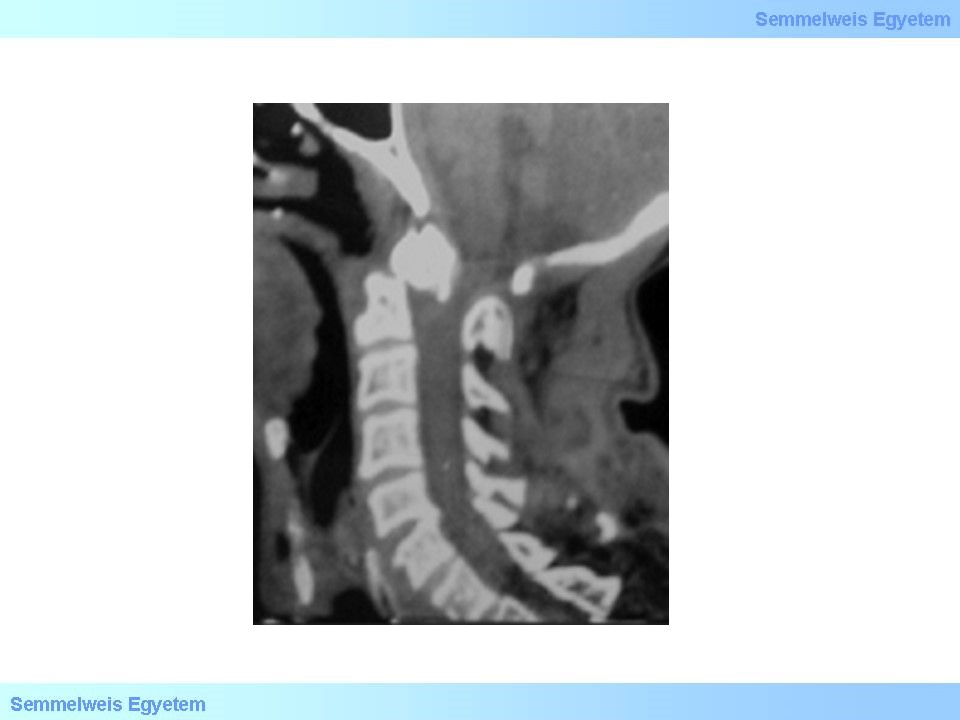
Keratán-szulfát lebontás zavara okozza a betegséget, amely kora gyermekkorban jelentkezik először és alacsonynövéssel, csontdeformitásokkal, corneahomállyal jár megtartott intellektus mellett. Az ízületek kóros mozgástartományban is mozgathatóak lesznek a szalagok lazasága miatt, így életveszélyes atlantoaxialis luxatio is kialakulhat gerincvelőkompresszióval (20. ábra) illetve a deformált mellkas miatt cor pulmonale chronicum (a chronicus hypoventillatio miatt).
|
ERT itt is rendelkezésre áll, a Vimizim (elosulfase-alfa) ígéretes lehetőségnek tűnik
|
|
ERT ebben a betegségben is rendelkezésre áll, a galsulfase (N-acetylgalactosamin- 4-sulfatase).
|
V./5.2.2.4. Mucopolysaccharidosis VI. (MPS VI., Maroteaux-Lamy-betegség)
A mucopolysaccharidosis VI. -ban szenvedők fenotípusos jegyei súlyos esetben a Hurler-szindrómásokkal nagy hasonlóságokat mutatnak, de intellektusuk megtartott és dermatán-szulfátot ürítenek a vizeletükkel. A tünetek széles spektrum mentén változhatnak, és a betegség progressziójával korrelálnak (22. ábra). A betegség első jelei kora gyermekkorban mutatkoznak, ha jellegzetes arcdysmorphiával nem is minden esetben jár, de szembetűnő a növekedésbeli elmaradásuk, a hepatomegalia (splenomegalia nem mindig alakul ki) és corneahomály is jellemző. Nagyon jellegzetes a testtartásuk a beszűkült ízületi mozgástartományok miatt (23. ábra).
V./5.2.3. Glicogenosisok, Pompe-kór
A szénhidrát anyagcsere betegségei esetében a szénhidrátok biokémiai átalakítása során következik be egy enzimatikus hiba. Ennek a csoportnak a legklasszikusabb példái a glikogén tárolási betegségek, azon belül is a Pompe-kór. Ez a betegség egyszersmind lysosomális tárolási betegség is, mivel a kialakulásáért felelés gén a lysosomális savas-alpha-glucosidase. A legtöbb enzimopathiához hasonlóan autoszomális recesszív öröklődést mutat. A enzim hiba súlyoságától és a visszamaradó enzimaktivitás mértékétől függően több súlyosági foka lehet a betegségnek. Gyermekkori indulású formáknál a cardiomyopathia dominálja klinikai képet, melyhez hypotonia, myopathia társul, továbbá a macroglossia is jellegzetes tünet. Felnőttkori típusban általában az alsó végtag myopathiája a vezető tünet, a progresszió lassabb, cardialis érintettség kevésbé jellemző. Ebben a betegségben is már régóta rendelkezésre áll enzimpótló terápia.
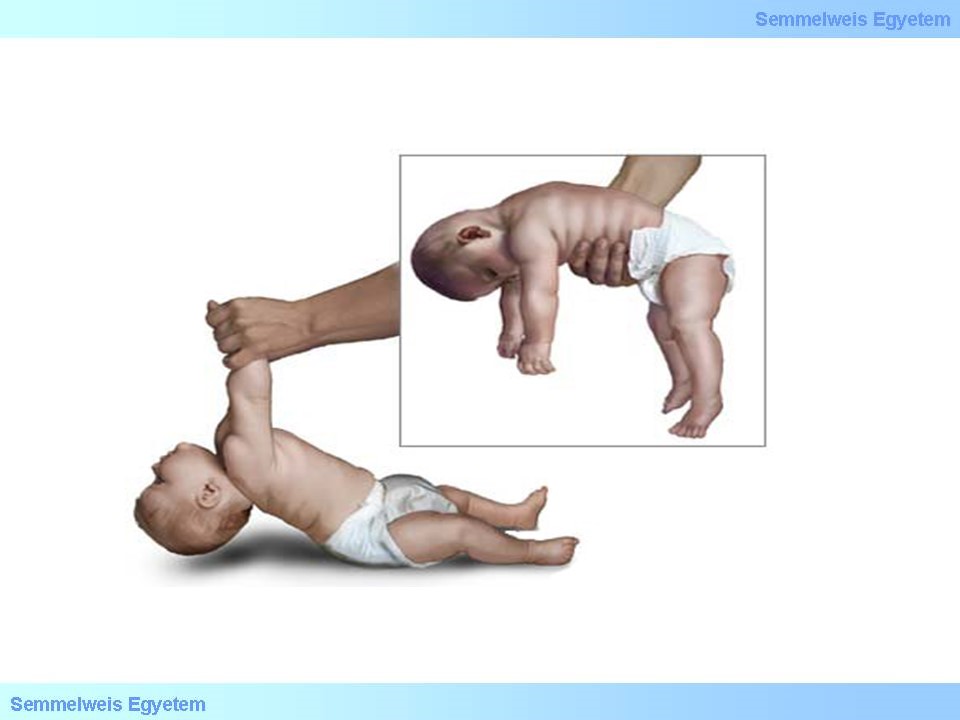
24. ábra: Floppy baby jel (hypotonia) Pompe-kór korai indulású formája esetén. Forrás: emaze.com
|

25. ábra: Módosított Gowers-jel (proximális izomgyengeség) a Pompe-kór késői indulású formájában Forrás: medscape.com
|
V./5.2.4. Glicoproteinosisok, Mannosidosis
A mannosidosisok a MAN2B1 gén mutációjaként alakulnak ki az α-mannozidáz enzim hiánya következményeként. Két formájukat különítjük el, az α-és β-mannosidosisokat, de a jelen fejezet terjedelemibe csak az α-mannosidosis tárgyalása fér bele. A klinikai tünetek itt is széles spektrumon belül változnak, de súlyos formában (mely kora gyermekkori indulású), ld. 26. ábra, a fenotípusos jellegek hasonlóságokat mutatnak a Hurler-kórral. Az elkülönítést segíti a kizárólag mannosidosisokban jellegzetes széles rés a metszőfogak között. A betegség legsúlyosabb formája az élet első hónapjaiban jelentkezik és gyors progressziójú. Csontrendszeri rendellenességekkel, mentalis retardatioval jár, myopathiaval is jár.
|
|